The Science of Mitophagy: Urolithin A’s Impact on Mitochondrial Function and Aging
Evaluating mitochondrial enhancing potential: Mitochondrial-enhancing compounds are evaluated based on their ability to improve mitochondrial efficiency by increasing ATP production and reducing oxidative stress. This dual effect supports cellular energy demands while minimizing oxidative damage, which is crucial for maintaining cellular health, especially in aging tissues where mitochondrial function declines.
Mitophagy as Mitochondrial Quality Control: Mitophagy is a specialized form of autophagy that selectively identifies and removes damaged mitochondria, ensuring optimal energy production (ATP) while reducing harmful byproducts like reactive oxygen species (ROS), which can lead to oxidative stress, aging, and diseases such as neurodegeneration and cardiovascular disorders.
PINK1/Parkin Pathway in Mitophagy: The PINK1/Parkin pathway is a key mechanism for identifying and removing damaged mitochondria. When a mitochondrion loses its membrane potential and becomes unable to produce ATP efficiently, PINK1 accumulates on its outer membrane, signaling dysfunction. PINK1 recruits Parkin, which tags the mitochondrion with ubiquitin, marking it for removal.
Recycling Damaged Mitochondria: Tagged mitochondria are engulfed by autophagosomes, which fuse with lysosomes to break down the damaged organelles into their basic components for recycling. This process is vital for cellular energy production and is disrupted in diseases like early-onset Parkinson's, highlighting its therapeutic potential.
The Importance of Mitophagy in High-Energy Tissues: Mitophagy is critical in tissues with high energy demands, such as the brain, heart, and muscles, where the removal of damaged mitochondria prevents oxidative stress and maintains ATP production. The age-related decline in mitophagy exacerbates mitochondrial dysfunction, contributing to chronic oxidative stress, neuronal death, and the progression of age-related diseases like Parkinson’s, Alzheimer’s, and cardiovascular conditions, highlighting its therapeutic potential.
Urolithin A as a Mitophagy-Activating Molecule: Urolithin A (UA), a metabolite derived from gut bacteria processing polyphenols in foods like pomegranates and walnuts, has emerged as a promising natural compound for enhancing mitophagy. By promoting the removal of damaged mitochondria and improving mitochondrial function, particularly in aging cells, UA supports healthy aging and has garnered significant scientific interest.
Role of Gut Health in Urolithin A Production: The synthesis of UA relies on specific beneficial gut bacteria, underscoring the importance of a balanced microbiome for its effective production and absorption. Without a healthy gut, the body's ability to access UA’s potential benefits, including its mitophagy-inducing effects, may be diminished.
Urolithin A and AMPK Activation: Urolithin A activates AMPK, a critical energy sensor in cells, to enhance mitophagy, restore mitochondrial health, and maintain energy balance, particularly in high-demand tissues like the brain, heart, and muscles. This AMPK-boosting effect is especially significant in aging, where diminished AMPK activity contributes to mitochondrial dysfunction and oxidative stress.
Urolithin A and mTOR Modulation: Urolithin A inhibits the overactive mTOR pathway in aging cells, relieving its blockade on autophagy and promoting mitophagy. By shifting cells from growth to maintenance mode, UA enables the removal of damaged mitochondria, reducing oxidative stress and enhancing mitochondrial function, thereby supporting a healthier and more resilient cellular environment.
Urolithin A and Autophagosome Formation: Urolithin A enhances autophagosome formation, a critical step in mitophagy where damaged mitochondria are enveloped and isolated for degradation. By activating pathways such as ULK1, UA ensures efficient tagging, engulfing, and recycling of defective mitochondria, promoting cellular efficiency and reducing oxidative damage, especially in aging cells.
Rapamycin and Mitophagy in Mitochondrial Disease: A 2022 study revealed that Rapamycin treatment (8mg/kg/day) increased mitophagy by 125% in a mitochondrial disease model, restoring hyperactivated autophagy states to normal levels through the reduction of mTOR activity and markers like phosphorylated s6 and P62, highlighting its potential to address mitochondrial quality control deficits.
Rapamycin's Role in Neurodegeneration: In Alzheimer's disease models, Rapamycin improved cognitive function, synaptic plasticity, and mitochondrial quality by increasing mitophagy markers such as TOM20 and LC3B co-localization, demonstrating its ability to enhance mitochondrial turnover and potentially mitigate age-related neuronal decline.
Urolithin A’s Role in Mitophagy and Inflammation: By enhancing mitophagy, Urolithin A selectively removes damaged mitochondria, reducing ROS production and mitigating inflammation. This dual action supports mitochondrial health, offering therapeutic potential for conditions like osteoarthritis, muscle weakness, and inflammatory diseases, while promoting both lifespan and healthspan.
Urolithin A Enhances Muscle Function and Endurance: Clinical studies demonstrate that Urolithin A supplementation significantly improves muscle strength, aerobic capacity, and endurance in both middle-aged and elderly adults, as evidenced by increased VO₂, longer 6-minute walk test distances, and improved muscle contractions before fatigue. These benefits are linked to enhanced mitochondrial function and the activation of mitophagy.
Systemic Benefits Through Reduced Inflammation and Enhanced Metabolism: Urolithin A reduces biomarkers of mitochondrial inefficiency and inflammation, such as acylcarnitines and C-reactive protein (CRP), indicating improved mitochondrial metabolism and decreased oxidative stress. These effects support sustained muscle performance and highlight its potential to combat age-related muscle decline.
Urolithin A and Metabolic Health: Urolithin A (UA) enhances insulin sensitivity, promoting efficient glucose uptake and reducing the risk of type 2 diabetes and metabolic disorders. Studies indicate that UA also decreases liver fat accumulation, shrinks fat cell size, and mitigates inflammation, addressing key risk factors for obesity and metabolic syndrome.
Cardiovascular Benefits of Urolithin A: Urolithin A (UA) enhances mitophagy in heart cells, replacing dysfunctional mitochondria to improve energy production, reduce oxidative stress, and enhance the heart’s contractile capacity. These effects support healthier blood vessel function and reduced risks of heart disease.
Clinical Insights into UA and Vascular Health: A 12-week study demonstrated that 50 mg of UA daily improved vascular endothelial function (VEF) and increased gut microbiota diversity, including beneficial bacteria linked to reduced cholesterol, better insulin sensitivity, and enhanced circadian rhythm regulation. These findings suggest a connection between UA’s cardiovascular and gut health benefits.
Health Benefits and Safety Profile of Urolithin A (UA): Urolithin A has demonstrated benefits in enhancing mitophagy, supporting muscle strength, neuroprotection, metabolic health, and cardiovascular function, with an established safety profile for doses between 250–1,000 mg daily. While short-term studies show minimal adverse effects, individual responses vary, and further research is needed to assess long-term safety and efficacy, particularly in diverse populations.
Background
Mitochondria are essential organelles responsible for generating adenosine triphosphate (ATP), which powers nearly all cellular functions, from muscle contraction to neural signaling. However, these essential organelles can become damaged or dysfunctional over time, leading to reduced energy output and an increase in harmful byproducts known as reactive oxygen species (ROS). While ROS play critical roles in cell signaling and immune responses, excessive levels lead to oxidative stress, damaging mitochondrial DNA (mtDNA), proteins, and lipids. This damage impairs mitochondrial function, creating a cycle of dysfunction that accelerates cellular aging and contributes to degenerative diseases such as neurodegeneration, cardiovascular disorders, and sarcopenia [1].
To mitigate mitochondrial damage, cells rely on a highly selective quality control mechanism known as mitophagy. This process identifies, removes, and replaces damaged mitochondria, ensuring optimal energy production and reducing oxidative stress. Mitophagy ensures optimal energy production and reduces oxidative stress, making it essential in tissues with high energy demands—such as the heart, brain, and skeletal muscles. However, as mitophagy efficiency declines with age, damaged mitochondria accumulate, leading to oxidative stress, inflammation, and tissue dysfunction. This impairment is strongly linked to the progression of age-related diseases, highlighting the critical role of maintaining mitochondrial quality in healthspan and longevity. [1]
Emerging research highlights the geroprotective potential of Urolithin A (UA), a gut-derived metabolite from dietary polyphenols found in foods like pomegranates and berries. Studies demonstrate that UA enhances mitophagy, promoting the efficient clearance of defective mitochondria, improving mitochondrial quality, reducing oxidative stress, and supporting healthy aging. These findings position UA as a promising intervention for addressing mitochondrial dysfunction, a hallmark of aging. By targeting mitochondrial health at its core, UA addresses foundational aspects of vitality and healthspan. [1]
In this review, we examine the science behind UA’s effects on mitophagy, its implications for age-related health, and its potential contribution to strategies aimed at promoting healthy aging and extending healthspan.
Mitophagy—Mitochondrial Quality Control Apparatus
The term "autophagy" has become a buzzword in health, longevity, and anti-aging circles. It refers to the process by which cells clean out damaged components, effectively renewing themselves. However, a less-discussed but equally important aspect is mitochondrial-specific autophagy, known as "mitophagy." Mitophagy operates much like traditional autophagy but focuses exclusively on identifying and removing damaged, dysfunctional, or inefficient mitochondria.
Mitophagy is a specialized form of autophagy—a cellular recycling process where cells degrade and renew their own components. In the case of mitophagy, the focus is on mitochondria, the tiny organelles often called the "powerhouses" of the cell. Mitochondria are essential because they convert the food we eat into adenosine triphosphate (ATP), the energy currency that powers nearly all cellular functions, from muscle contraction to nerve impulse transmission. However, mitochondria are not immune to damage. Stressors such as toxins, oxidative damage, or the natural wear and tear of metabolic processes can impair their function over time.
Think of mitochondria as the engines of a car. When they're running smoothly, they efficiently burn fuel to produce energy. But if these engines start to malfunction—perhaps due to poor maintenance or faulty parts—they become less efficient at producing energy (ATP) and begin to emit more exhaust fumes. The "exhaust fumes" are excessive amounts of reactive oxygen species (ROS), which are highly reactive molecules formed as byproducts of oxygen-dependent processes within mitochondria.
While ROS at normal levels play important roles in cell signaling and immune responses, too much "exhaust" can be corrosive to the cellular environment. This overproduction leads to oxidative stress, a condition where the balance between ROS and the body's ability to neutralize them is disrupted. Over time, this cumulative damage accelerates aging and contributes to various diseases, including neurodegenerative disorders like Alzheimer's and Parkinson's disease, cardiovascular ailments, and certain cancers.
Evaluating the efficacy of mitochondrial-enhancing compounds requires an understanding of their impact on mitochondrial efficiency. Specifically, such compounds should demonstrate the ability to increase ATP production while simultaneously reducing oxidative stress. Enhanced mitochondrial efficiency translates to improved energy output for cellular processes and minimizes the harmful byproducts that contribute to oxidative damage. These outcomes are critical for maintaining cellular health, particularly in aging tissues where mitochondrial function naturally declines.
Mitophagy plays a vital role in preventing this cascade of damage. By identifying and selectively removing defective mitochondria, mitophagy preserves mitochondrial efficiency and reduces the production of excessive ROS. This process ensures that only healthy mitochondria remain, thereby supporting sustained energy production and minimizing the risks of oxidative damage. In tissues with high energy demands, such as the brain, heart, and skeletal muscles, efficient mitophagy is particularly crucial for maintaining cellular and systemic health. [1]
To understand the critical role of mitophagy in maintaining mitochondrial health, it is essential to examine how cells identify dysfunctional mitochondria and orchestrate their removal. The process is highly regulated, involving a series of molecular signals and pathways that ensure only damaged mitochondria are targeted while preserving healthy ones. In the following section, we delve into the mechanisms underlying mitophagy, focusing on the cellular machinery responsible for recognizing, tagging, and ultimately recycling compromised mitochondria to maintain energy balance and prevent oxidative damage.
How Cells Identify and Recycle Damaged Mitochondria
One of the most well-understood mechanisms of mitophagy involves the PINK1/Parkin pathway. PINK1, which stands for PTEN-induced kinase 1, is a protein that typically gets imported into healthy mitochondria and is rapidly broken down. However, when a mitochondrion becomes damaged—specifically when its membrane potential drops, indicating a loss of the energy gradient across the mitochondrial membrane—PINK1 accumulates on the outer membrane instead of degrading. [1]
This drop in membrane potential signals that the mitochondrion is no longer efficiently processing electrons through the electron transport chain to generate ATP. In healthy mitochondria, a strong membrane potential is maintained by the electron transport chain, which generates ATP by creating an energy gradient across the membrane. This gradient is essential for energy production, as it drives the synthesis of ATP, the cell’s primary energy currency. When the membrane potential falls, the mitochondrion loses its ability to produce ATP efficiently, often leading to the accumulation of reactive byproducts (ROS) that can damage other cellular structures. The loss of membrane potential essentially marks the mitochondrion as compromised and unable to meet the cell’s energy demands. [1]
The accumulation of PINK1 on the outer mitochondrial membrane acts like a distress beacon, signaling that something is amiss. PINK1 then recruits another protein called Parkin to the damaged site. Parkin is an enzyme that attaches small molecules called ubiquitin to proteins on the mitochondrial surface. This process, known as ubiquitination, effectively tags the mitochondrion for removal. [1]
Once marked, the defective mitochondrion is enveloped by an autophagosome—a cellular "container" with a double membrane designed to isolate and transport cellular debris. The autophagosome then fuses with a lysosome, an organelle filled with digestive enzymes. Inside the lysosome, the damaged mitochondrion is broken down into its basic components. These components can then be recycled to build new mitochondria or utilized in other cellular functions. [1]
Understanding this pathway is crucial because disruptions in the PINK1/Parkin mechanism have been linked to several neurodegenerative diseases. For instance, mutations in the genes encoding PINK1 or Parkin are associated with early-onset Parkinson's disease. Enhancing or mimicking this pathway could offer therapeutic avenues for conditions caused by mitochondrial dysfunction.
Mitophagy and Aging
As you can imagine, mitophagy is crucial in tissues with high energy demands, such as the heart, brain, and muscles. In these tissues, cells depend on a constant supply of ATP to function correctly. If damaged mitochondria are not removed, they fail to produce sufficient energy and contribute to increased ROS levels, leading to the corrosion of the cell itself and ultimately tissue damage. [1]
Adding to the vulnerability, mitochondria contain their own DNA (mtDNA), which is separate from the cell’s nuclear DNA. Unlike nuclear DNA, mtDNA is not as well-protected and is more susceptible to damage from oxidative stress. When ROS accumulate, they can harm mtDNA, leading to mutations that impair mitochondrial function. Damaged mtDNA not only reduces ATP production but also impairs the mitochondria’s own mechanisms for self-repair and replication, creating a vicious cycle of dysfunction and mitochondrial decline. Over time, this mtDNA damage can contribute to aging and the onset of age-related diseases, including neurodegenerative conditions like Alzheimer's and Parkinson’s, as well as cardiovascular diseases where the energy needs of tissues are high and must remain stable. [1]
At the same time, as we age, the efficiency of mitophagy declines. Cells become less adept at recognizing and removing dysfunctional mitochondria, allowing these damaged mitochondria to accumulate within tissues over time. Returning to the metabolic engine analogy, our cellular engines gradually become highly inefficient in generating energy. Just as a poorly maintained engine produces more exhaust, these malfunctioning mitochondria emit excessive "metabolic exhaust" in the form of ROS.
This excess ROS leads to chronic oxidative stress—a hallmark of aging—that damages critical cellular structures. Over time, this oxidative damage contributes to a progressive decline in cellular function and is implicated in a variety of age-related diseases, such as neurodegenerative disorders like Parkinson's and Alzheimer's, as well as cardiovascular conditions that depend on optimal energy metabolism. [1]
In neurodegenerative diseases, impaired mitophagy is closely linked to neuronal death. Neurons, unlike many other cell types, are largely non-replicative, meaning they cannot be readily replaced once lost. This makes them especially vulnerable to accumulated damage.
In Parkinson’s disease, for example, the PINK1/Parkin pathway—a key mechanism in mitophagy—frequently becomes defective. Mutations in the genes for PINK1 or Parkin disrupt the cell’s ability to clear damaged mitochondria, leading to a buildup of dysfunctional mitochondria that contribute to oxidative stress and, ultimately, the death of dopamine-producing neurons. This neuronal loss is what drives the hallmark motor symptoms of Parkinson’s disease, such as tremors, rigidity, and bradykinesia (slowness of movement). [1]
This age-related decline in mitophagy not only highlights the importance of this process in maintaining cellular health but also underscores the potential therapeutic value of enhancing mitophagy. By targeting mitophagy pathways, researchers hope to slow down or even reverse aspects of cellular aging, reduce the burden of damaged mitochondria, and help prevent the onset of conditions tied to mitochondrial dysfunction.
Can We Manipulate Mitophagy?
Emerging evidence suggests that enhancing mitophagy can positively impact longevity and lifespan. Several studies involving genetic mutation, dietary manipulations, or pharmacological interventions have demonstrated that promoting mitophagy extends the lifespan in various model organisms, including yeast [14], worms [15], flies, and mice [16].
Enhancing mitophagy through pharmacological interventions or lifestyle modifications holds promise for mitigating age-related diseases and promoting healthy aging. Here we will briefly discuss how geroprotective pharmacological interventions can improve mitophagic regulation, improve mitochondrial quality control as we age, and prevent the accumulation of damaged mitochondria within cells & tissues.
Urolithin A and Mitophagy
Given mitophagy's central role in aging and disease, researchers are actively investigating ways to enhance this process to promote healthy aging. One promising compound is Urolithin A (UA), a metabolite produced in the gut from digesting certain polyphenols found in foods like pomegranates. Emerging preclinical and human studies are showing compelling data on UA’s ability to stimulate mitophagy, drawing significant interest from scientists for its potential as a natural mitophagy-activating molecule. UA has been shown to activate mitophagy, promoting the removal of damaged mitochondria and improving mitochondrial function, particularly in aging cells where this naturally declines. [1]
Urolithin A is not produced directly by our bodies but is instead synthesized by certain beneficial bacteria in the gut. When we consume foods rich in ellagitannins—a type of polyphenolic compound present in pomegranates, walnuts, and various berries—these compounds are converted by gut bacteria into urolithins, including UA. This conversion highlights the importance of gut health and a balanced microbiome, as specific bacteria are required to produce UA effectively. Without a healthy microbiome, the body’s ability to generate and absorb UA from diet may be reduced, underscoring how critical gut health is in accessing UA’s potential benefits. [1]
Chemically, UA is a small molecule composed of carbon, hydrogen, and oxygen atoms. You can think of it as a house with multiple rooms (the "rooms" representing different molecule parts). Its core structure consists of two interconnected rings, which play an essential role in its biological activity. Hydroxyl groups (-OH) are attached to this structure, like arms sticking out from the molecule. These hydroxyl groups enable UA to interact with various enzymes and molecules in the body, much like a key fitting into a lock. This structure is critical for UA's ability to induce mitophagy by fitting into and activating the cellular pathways responsible for clearing out damaged mitochondria. [1]
The exact mechanism by which UA promotes mitophagy is complex and involves multiple steps, which will be further elaborated upon in the following sections. Urolithin A promotes mitophagy through several interconnected mechanisms that are crucial for maintaining cellular health, particularly in aging cells.
Activation of AMPK
Urolithin A activates AMPK (AMP-activated protein kinase), an essential energy sensor and regulator in cells. AMPK plays a critical role in maintaining cellular energy balance, acting much like an internal "fuel gauge." When energy levels within the cell are low—such as during periods of nutrient scarcity or increased energy demand—AMPK is activated to restore balance. This activation prompts cells to increase energy production and limit energy-consuming processes, a response critical for cellular survival and function.
In addition to its role as an energy sensor, AMPK also responds to mitochondrial stress. When mitochondria become dysfunctional, they can no longer efficiently produce ATP, leading to an energy deficit in the cell. By activating AMPK, cells initiate a cascade of protective mechanisms designed to restore mitochondrial health and energy balance. One of these mechanisms is mitophagy.
Urolithin A’s activation of AMPK signals the cell to enhance mitophagy, thereby targeting malfunctioning mitochondria for removal. This process ensures that only healthy, functional mitochondria are maintained, which is crucial for cells with high energy demands, such as those in the brain, heart, and muscles. By supporting AMPK activation, UA helps to maintain cellular energy equilibrium, reducing oxidative stress and preventing the accumulation of damaged mitochondria that could otherwise compromise cell function [1].
This AMPK-boosting effect of UA has captured scientific interest due to its potential for addressing age-related declines in mitochondrial function. As we age, AMPK activity tends to decrease, diminishing the cell’s ability to maintain energy balance and increasing vulnerability to oxidative damage and cellular dysfunction. UA’s ability to activate AMPK is one of the ways it helps maintain mitochondrial integrity as we age [1].
Inhibition of mTOR
In addition to activating AMPK, Urolithin A also inhibits the mTOR (mammalian target of rapamycin) pathway, a critical regulator of cell growth, proliferation, and metabolism. The mTOR pathway acts as a cellular nutrient sensor, promoting anabolic processes—such as protein synthesis and cell growth—when nutrients and energy are abundant. However, with aging, mTOR activity often becomes excessively elevated, which can have detrimental effects on cellular health. An overactive mTOR pathway inhibits autophagy, including mitophagy, the essential processes that enable cells to remove damaged organelles and maintain homeostasis [1].
Under normal conditions, mTOR is beneficial when cells need to build and grow, as it stimulates protein synthesis, lipid production, and other growth-promoting processes. Prolonged mTOR activation with age can prevent cells from entering autophagy, a cellular "self-cleaning" state that breaks down damaged components, including mitochondria. This inhibition of autophagy leads to their accumulation, causing increased oxidative stress, reduced cellular efficiency, and contributing to age-related cellular decline.
Urolithin A helps to modulate this balance by inhibiting the mTOR pathway, thereby alleviating its blockage on autophagy and allowing mitophagy to proceed. By dampening mTOR activity, UA essentially shifts the cell from a growth mode to a maintenance mode, prioritizing the removal of damaged mitochondria over growth processes. This shift is vital for cellular health, as it enables the recycling and replacement of defective mitochondria, reducing oxidative damage and promoting more efficient energy production. In aging cells, where dysfunctional mitochondria tend to accumulate, Urolithin A’s inhibition of mTOR supports a "cleaner" and more resilient cellular environment.
Urolithin A’s dual action—activating AMPK and inhibiting mTOR—targets key pathways that regulate energy balance, growth, and maintenance. By encouraging mitophagy through both pathways, UA provides a powerful strategy for promoting mitochondrial health, slowing cellular aging, and potentially reducing the risk of age-related diseases associated with mitochondrial dysfunction. [1]
Expression of Autophagy-Related Genes (PINK1/Parkin Pathway)
Urolithin A also directly influences the PINK1/Parkin pathway, a critical component of mitophagy. As we’ve discussed, PINK1 (PTEN-induced kinase 1) accumulates on the outer membrane of damaged mitochondria, where it acts as a distress signal, flagging these mitochondria for removal. This accumulation of PINK1 on damaged mitochondria signals that they are no longer functioning optimally. In response, PINK1 recruits Parkin, an enzyme that attaches ubiquitin molecules to the damaged mitochondria, effectively tagging them for destruction. This ubiquitination process marks the mitochondria for encapsulation and degradation within autophagosomes, which subsequently fuse with lysosomes to break down and recycle the mitochondrial components. [1]
Urolithin A has been shown to enhance this critical pathway by upregulating the expression of both PINK1 and Parkin, increasing the levels of these proteins within the cell. By boosting PINK1 and Parkin availability, Urolithin A ensures that malfunctioning mitochondria are quickly identified, tagged, and directed toward the mitophagy pathway for removal. This effect is especially important in aging cells, where reduced mitophagy efficiency can lead to the accumulation of damaged mitochondria. Such accumulations contribute to cellular dysfunction and oxidative stress, which are key factors in aging and age-related diseases. [1]
Formation of Autophagosomes
Once Parkin tags the damaged mitochondria for degradation, they are enveloped by autophagosomes—specialized cellular structures that function as waste collectors. The autophagosome wraps the damaged mitochondria in a double membrane, effectively isolating them from the rest of the cell and ensuring that the breakdown products don’t harm surrounding cellular components.
Urolithin A promotes this essential step in mitophagy by enhancing the formation of autophagosomes. When AMPK is activated, as we’ve seen, it initiates a signaling cascade that modifies a key protein called ULK1 (Unc-51 Like Autophagy Activating Kinase 1). This modification is crucial, as ULK1 is a primary regulator in the early stages of autophagosome formation. ULK1 helps form structures called phagophores, which are the initial, cup-shaped membranes that mature into autophagosomes. Phagophores grow and curve around the damaged mitochondria, eventually closing off to form a fully enclosed autophagosome capable of engulfing and isolating malfunctioning mitochondria. [1]
Once the autophagosome is fully formed around the tagged mitochondria, it fuses with lysosomes, which are organelles packed with digestive enzymes. This fusion allows the lysosomal enzymes to break down the damaged mitochondria into their basic components, which are subsequently recycled by the cell for other purposes. Through this recycling process, the cell maintains a pool of usable molecules, effectively conserving resources and ensuring cellular efficiency.
Urolithin A’s support of autophagosome formation highlights its comprehensive role in promoting cellular health. By enhancing both the initiation of mitophagy and the clearance of defective mitochondria, Urolithin A contributes to a cellular environment that is less prone to oxidative damage and more resilient in energy production. This complete cycle—from tagging to engulfing and recycling—illustrates why mitophagy is essential for long-term cellular health and how Urolithin A aids in maintaining mitochondrial quality, especially in aging cells where such processes typically slow down. [1]
The Comprehensive Process of Urolithin A-Induced Mitophagy
The process by which Urolithin A promotes mitophagy can be summarized in four key steps:
- AMPK activation: Urolithin A activates AMPK, which senses the need for energy balance and signals the initiation of mitophagy to clear damaged mitochondria.
- Inhibition of mTOR: Urolithin A inhibits the mTOR pathway, which is often overactive in aging cells. This removes the block on autophagy and allows the cell to clear out waste like damaged mitochondria.
- Upregulation of PINK1/Parkin: Urolithin A increases the expression of PINK1 and Parkin proteins, essential for identifying and tagging damaged mitochondria for removal.
- Formation of Autophagosomes: Urolithin A promotes the formation of autophagosomes by activating ULK1, initiating the creation of phagophores that mature into autophagosomes. These structures engulf the damaged mitochondria and fuse with lysosomes, leading to the breakdown and recycling of mitochondrial components.
These steps ensure that cells can efficiently remove dysfunctional mitochondria, preventing the buildup of cellular debris that leads to oxidative stress and accelerates aging. By keeping this process intact, Urolithin A supports healthier mitochondrial function, which is essential for long-term cellular health and longevity.
Scientific Evidence Supporting Urolithin A and Mitophagy
Preclinical Animal Studies
A growing body of evidence supports the connection between Urolithin A and its ability to stimulate mitophagy. For instance, a study by Luan et al. (2021) investigated Urolithin A’s potential as a therapeutic approach for Duchenne muscular dystrophy (DMD), a severe genetic disorder primarily affecting boys. DMD leads to progressive muscle weakness and degeneration due to mutations in the dystrophin gene, which is crucial for maintaining muscle cell structure and function. Dystrophin acts as a stabilizing protein, linking the muscle cell membrane to the extracellular matrix and enabling muscles to withstand repeated contractions. In the absence of dystrophin, muscle cells become fragile, weakening over time and progressively deteriorating.
One of the key challenges in DMD is mitochondrial dysfunction, which significantly contributes to the disease’s progression. Mitochondria in muscle cells affected by DMD exhibit reduced functionality, lower energy production, and increased oxidative stress. Furthermore, research has shown that genes involved in mitophagy are often underexpressed in DMD, reducing the muscle cells' ability to clear out damaged mitochondria effectively. Studies on DMD mouse models have consistently revealed decreased levels of mitophagy markers, indicating impaired mitophagy in this disease context. [2]
The researchers in this study explored whether Urolithin A could activate the mitophagy process to address this issue. They tested the effects of Urolithin A on both mice and human muscle cells. In the mice, Urolithin A was added to their food at a dose of 50 mg per kg per day. In human trials, muscle cells were obtained from three healthy boys and three boys with DMD, all aged 4 to 7 years. These cells were treated with 25 μM of Urolithin A for varying durations (2, 6, and 24 hours). The study found that Urolithin A enhanced the clearance of defective mitochondria in both the mice and the DMD-affected human muscle cells. This improvement led to better mitochondrial function and increased the muscle's ability to use oxygen, suggesting that Urolithin A may reduce symptoms of DMD by promoting mitophagy. [2]
A recent study by Huang et al. (2023), titled Urolithin A Ameliorates Obesity-Induced Metabolic Cardiomyopathy in Mice via Mitophagy Activation, published in the prestigious journal Nature, explored Urolithin A’s role in managing metabolic cardiomyopathy (MC), a condition often associated with obesity. MC is characterized by the buildup of fat within heart cells, which disrupts normal cardiac function and leads to oxidative stress and mitochondrial dysfunction. This condition is particularly concerning because it impairs the heart's energy production, weakens contractility, and lacks effective treatment options.
In cases of obesity, cardiomyocytes (heart muscle cells) exhibit distinct mitochondrial abnormalities. Mitochondria in these cells often show impaired respiration, mislocalization within the cell, and disorganized mitophagy—a process critical for mitochondrial quality control. Mitochondria undergo constant dynamic cycles, including fission (division), fusion (joining), biogenesis (creation of new mitochondria), and mitophagy (selective degradation). Among these processes, mitophagy plays a key role in maintaining mitochondrial homeostasis by removing damaged or aged mitochondria, thus preventing the accumulation of dysfunctional organelles that could compromise cellular health.
In healthy cells, mitophagy is facilitated by pathways like the PINK1/Parkin or receptor-dependent pathways, both of which target damaged mitochondria for degradation and recycling. However, in the cardiomyocytes of MC mice, researchers observed enlarged mitochondria with collapsed membrane potentials, indicating severe dysfunction. These mitochondria, which are unable to maintain the necessary energy gradient, were not being cleared effectively, suggesting that impaired mitophagy was contributing to the condition. This buildup of damaged mitochondria likely exacerbates oxidative stress and further weakens mitochondrial function. [3]
In this study, Huang et al. sought to determine if poor mitophagy is a driving factor in MC and whether Urolithin A could mitigate these effects. Using an obesity-induced MC model in mice, they fed the mice a high-fat diet for 20 weeks to induce obesity and MC symptoms, followed by a 4-week Urolithin A treatment at a dosage of 50 mg per kg per day. The aim was to assess whether Urolithin A could restore mitophagy, thereby reducing mitochondrial dysfunction and improving cardiac health. [3]
The researchers found a strong link between obesity and reduced mitophagy in heart cells. In the Urolithin A-treated mice, mitophagy activity significantly improved, and symptoms of MC were reduced. When heart cells were exposed to palmitic acid (a fat), 5 μM of Urolithin A increased the production of autophagosomes (which engulf damaged mitochondria) while reducing autolysosomes (which break down autophagosomes). Urolithin A helped balance these processes, allowing better mitochondrial cleaning. Moreover, when mitophagy blockers or gene silencing techniques were used to inhibit Parkin (a key protein in mitophagy), the protective effects of Urolithin A were reduced, further proving that Urolithin A's benefits were tied to restoring mitophagy. [3]
These animal studies show that Urolithin A enhances mitophagy, improving conditions like DMD and metabolic cardiomyopathy by promoting mitochondrial function and reducing disease progression.
Clinical Studies
In addition to preclinical animal studies, research on Urolithin A has been extended to human trials. A study by Singh et al. (2022), titled UA Improves Muscle Strength, Exercise Performance, and Biomarkers of Mitochondrial Health in a Randomized Trial in Middle-Aged Adults, investigated the effects of Urolithin A on mitophagy in middle-aged adults over a period of four months. [4]
The Study Design
The study sought to determine the most responsive functional outcomes, specifically focusing on markers linked to muscle strength, exercise tolerance, and overall physical performance. The goal was to identify outcomes that could guide the design and power calculations of future confirmatory clinical trials involving UA.
The study population included 88 untrained adults between the ages of 40 and 64 who were overweight and demonstrated low physical endurance, defined as a maximum oxygen consumption (VO2max) below 35 mL/kg/min. Participants were selected from an initial screening of 253 individuals based on inclusion and exclusion criteria, which required that subjects be healthy, as determined by their vital signs, anthropometric measures, and absence of chronic medical conditions. Participants were randomized into three groups: a 500 mg UA group, a 1,000 mg UA group, and a placebo group. The intervention spanned a 4-month period, chosen as the minimum duration expected to reveal impacts on physical performance and muscle function according to guidelines from expert groups on muscle-function clinical trials.
The ATLAS study was a double-blind, placebo-controlled trial with well-matched baseline characteristics across groups. Participants in the 500 mg UA, 1,000 mg UA, and placebo groups had similar average ages (around 51–54 years), BMIs (approximately 29 kg/m²), and baseline endurance (VO2max around 23 mL/kg/min). The study cohort included a higher proportion of female participants (2:1 female-to-male ratio) and was primarily of Western European ethnicity.
Throughout the study, plasma samples were collected to assess UA’s impact on metabolites and cytokines associated with cellular health, while skeletal muscle biopsies were performed to evaluate UA’s effects on the muscle transcriptome and proteome, with a specific focus on proteins related to mitochondrial health and cellular energy pathways. This comprehensive approach was designed to yield insights into UA’s potential to influence cellular health markers, muscle function, and overall physical performance.
Results
The study showed that participants who received Urolithin A exhibited enhanced mitophagy, significantly improving muscle strength (around 12%) and aerobic endurance. Participants receiving 500 mg and 1,000 mg UA exhibited notable increases in hamstring muscle strength, measured via isokinetic Biodex dynamometer testing. The average peak torque for hamstring muscles increased significantly in both groups, with a 12% increase in the 500 mg group (p = 0.027) and a 9.8% increase in the 1,000 mg group (p = 0.029) compared to placebo. Maximum torque during knee flexion also improved significantly in both UA groups, with increases of 10.6% in the 500 mg group (p = 0.017) and 10.5% in the 1,000 mg group (p = 0.022) compared to placebo.
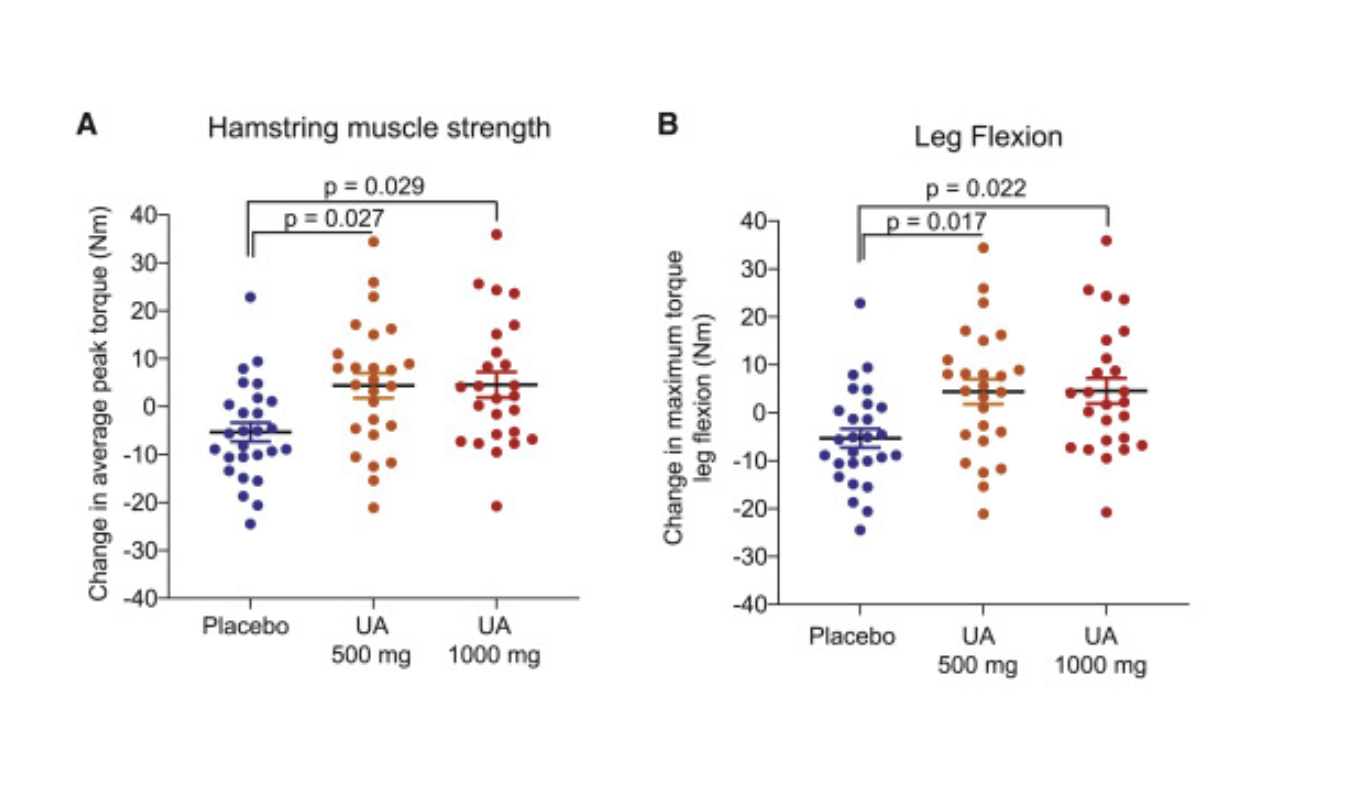
Aerobic endurance was measured by peak oxygen consumption (VO₂), which indicates how efficiently the body uses oxygen during exercise, and the 6-minute walk test (6MWT), a simple measure of physical endurance. [4]
PPO was the study’s primary pre-specified endpoint. Although no significant differences were observed between the UA-supplemented groups and the placebo group, both UA groups demonstrated non-significant increases in PPO of approximately 4% from baseline, suggesting a trend towards improved power output. The placebo group, in contrast, showed no change in PPO, indicating that UA supplementation may support small gains in exercise performance that require further investigation to confirm statistical significance.
Participants in the 1,000 mg UA group exhibited a 15% increase in total cycling distance from baseline to the end of the study (p = 0.03), along with a significant increase in time to fatigue during the exercise test. This finding indicates that UA at a higher dose may contribute to improved endurance, allowing participants to cycle further and sustain activity longer before fatigue.
The UA 1,000 mg dose group showed significant within-group increases in peak VO₂ and estimated VO₂max at both the 2-month midpoint and 4-month endpoint (p < 0.01), demonstrating a measurable improvement in aerobic endurance. Although these changes were not statistically significant compared to the placebo, they showed a strong trend in favor of the UA intervention (p = 0.058), particularly in the 1,000 mg dose group.
Interestingly, no significant changes in acylcarnitine levels were observed in the 1,000 mg UA group. This absence of change suggests that the downregulation of acylcarnitines may be dose- or duration-dependent, with the lower dose potentially providing an optimal level of UA for impacting this specific biomarker. It is also possible that the observed reductions in the 500 mg group reflect a response that emerges over time and may require a balance in dosing to affect certain plasma biomarkers.
The study then analyzed the overall effects on inflammation and mitochondrial function. To determine if the improvements in muscle function observed with UA supplementation were also reflected in surrogate plasma biomarkers of metabolic health, researchers analyzed acylcarnitine levels in participants’ plasma. Acylcarnitines are lipid molecules involved in fatty acid metabolism, and reductions in acylcarnitine levels, particularly in medium- and long-chain species, are often associated with enhanced fatty acid oxidation and metabolic efficiency.
In the UA 500 mg group, plasma acylcarnitine levels were reduced, with the most significant decreases seen in medium- and long-chain acylcarnitines. This downregulation suggests an improvement in fatty acid oxidation, as lower levels of circulating acylcarnitines indicate that fatty acids are being more effectively broken down and utilized as energy within cells. This finding aligns with prior clinical studies that have linked acylcarnitine reduction to improved mitochondrial function and metabolic health
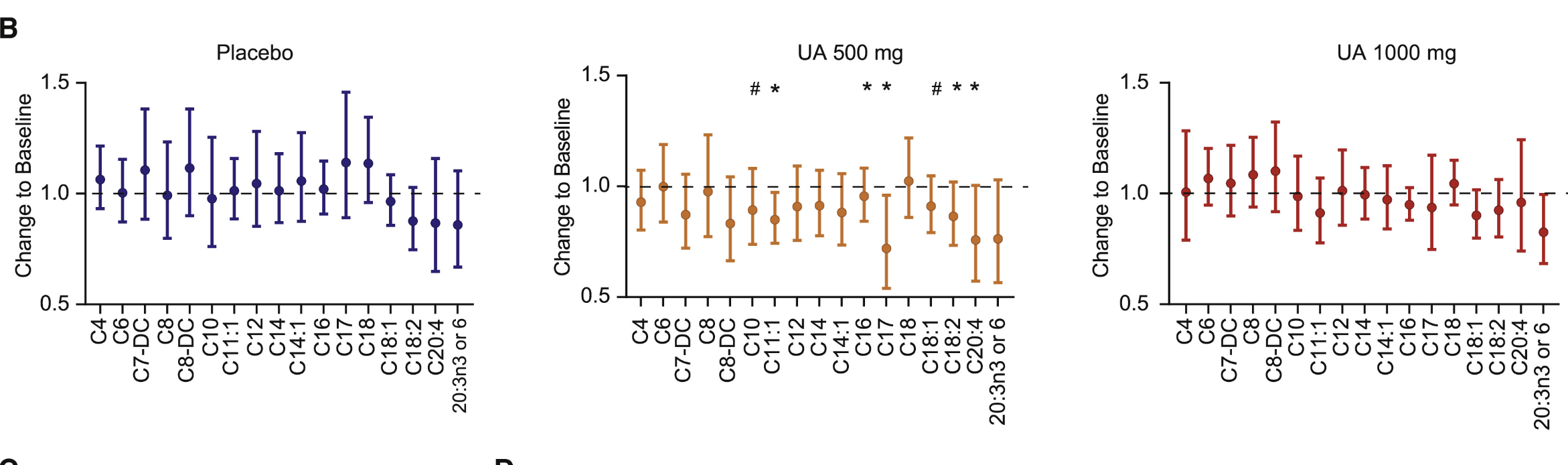
In addition to mitochondrial efficiency, the study also analyzed markers of inflammation. Inflammation is a key factor in aging and metabolic dysfunction, making it a critical area of focus for assessing the broader benefits of UA supplementation. This study evaluated plasma levels of C-reactive protein (CRP) and several pro-inflammatory cytokines to determine whether UA’s effects on muscle function and mitochondrial health are accompanied by systemic anti-inflammatory benefits.
CRP is a well-established biomarker of systemic inflammation and a predictor of age-related chronic diseases. Elevated CRP levels are common in overweight individuals and are strongly associated with higher BMI. At baseline, the study population of middle-aged, overweight participants had average CRP concentrations of approximately 3 mg/L, placing them in a moderate- to high-risk category for chronic disease.
UA supplementation reduced plasma CRP levels at both doses, with the reduction reaching statistical significance in the 1,000 mg group. This suggests that UA at higher doses may effectively lower systemic inflammation, potentially mitigating risks associated with chronic inflammatory states.
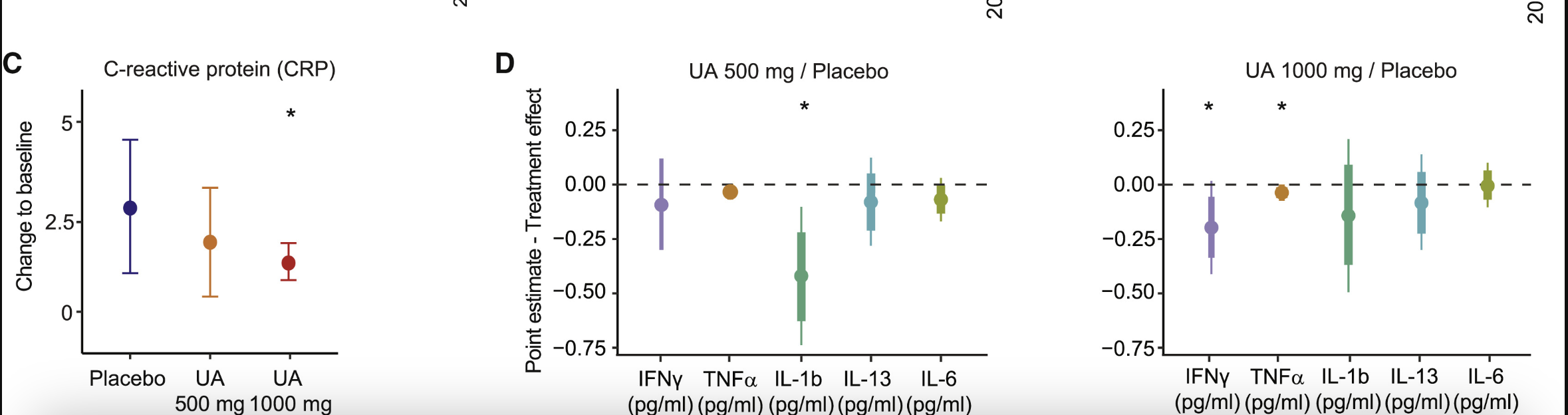
In addition to CRP, UA supplementation led to reductions in key pro-inflammatory cytokines, including interferon gamma (IFN-γ), interleukin-1 beta (IL-1β), and tumor necrosis factor alpha (TNF-α). These cytokines are major mediators of inflammatory processes and are often elevated in chronic inflammatory and age-related diseases. Although baseline cytokine levels in this study population were relatively low, reductions were observed following UA supplementation, suggesting a mild systemic anti-inflammatory effect.
The decrease in CRP suggests that Urolithin A also helped reduce systemic inflammation, likely due to its enhancement of mitophagy and mitochondrial metabolism in skeletal muscle. [4]
These findings suggest that Urolithin A contributes to better mitochondrial function, improved aerobic capacity, and reduced inflammation in middle-aged adults, making it a promising intervention for combating muscle decline with age.
Another study by D'Amico et al. (2022), titled Urolithin A Improves Mitochondrial Health, Reduces Cartilage Degeneration, and Alleviates Pain in Osteoarthritis, explored the potential of Urolithin A in treating osteoarthritis (OA), a common joint disorder with no effective therapy. OA affects approximately 500 million people globally, leading to cartilage degradation, pain, and impaired mobility. Mitochondrial dysfunction is known to contribute to OA progression. The study examined the effects of Urolithin A on human knee joint cells from both healthy donors and OA patients, showing that Urolithin A improved mitophagy and mitochondrial respiration in joint cells. [5]
In a mouse model of OA, Urolithin A reduced disease progression by decreasing cartilage degradation, synovial inflammation, and pain. Furthermore, increased mitophagy and mitochondrial content in the joints of OA mice were associated with slower disease progression, suggesting that Urolithin A's ability to enhance mitophagy could offer therapeutic benefits in OA management. [5]
The collective findings from animal and human studies underscore the therapeutic potential of Urolithin A in promoting mitophagy and improving mitochondrial health. In studies like Singh et al. (2022) and D'Amico et al. (2022), Urolithin A enhanced mitophagy, improved muscle health and aerobic endurance, and even helped reduce inflammation and joint degradation in osteoarthritis. These studies suggest that Urolithin A holds promise as a therapeutic intervention for managing age-related conditions like muscle decline and joint disorders by targeting mitochondrial dysfunction.
Comparative Analysis of Urolithin A vs. Other Longevity Protocols
Urolithin A (UA) distinguishes itself among longevity-promoting interventions due to its unique ability to specifically enhance mitochondrial quality control. While many compounds and protocols focus broadly on inducing autophagy—the cellular process that removes damaged or dysfunctional components—UA’s targeted action on mitophagy gives it a distinct advantage in improving mitochondrial health. Mitophagy, as previously discussed, focuses exclusively on the selective clearance of damaged mitochondria, the energy-producing organelles essential for cellular function and metabolic health.
Unlike general autophagy-inducing compounds, UA’s specificity for mitophagy directly addresses the mitochondrial dysfunction that underpins many aspects of aging and chronic disease. Below, we explore how UA compares to other longevity protocols and why its targeted mechanism makes it a promising candidate for improving mitochondrial health and overall cellular function.
Rapamycin and Mitophagy
It's well established that Rapamycin induces autophagy by suppressing mTOR activity. Therefore, it should likely have an effect on mitochondrial-specific autophagy, right?
Surprisingly, there's not too much evidence on the effects of Rapamycin on mitophagy. However, a recent 2022 paper published in Cell Metabolism by Dr Tom McWilliams has characterized the role of Rapamycin in mitophagy in a model of mitochondrial disease using the previously mentioned reporter models, which he developed during his PhD [17]. This model has some similarities to aging, in which older adults, especially octogenarians >80yrs age, display severe mitochondrial abnormalities [18].
The study shines light on the role of this powerful drug on mitochondrial quality control. The model of mitochondrial disease typically displays respiratory chain defects, 'ragged-red fibers' (expressing pathogenic mitochondria DNA variants & severe accumulation of damaged mitochondria) [17].
This study showed that the treatment of Rapamycin at a dose of 8mg/kg/day for 70 days resulted in a 125% increase in mitophagy [17]. The authors speculated that in aging and mitochondrial disease, there's hyperactivation of autophagy-mediated degradation as indicated by the accumulation of P62 and phosphorylated ubiquitin in muscle fibers [17]. Notably, compared to healthy control patient samples, patients with mitochondrial disease displayed an >80-fold increase in the autophagy adapter protein P62, with rapamycin treatment causing a ~90% decrease in the hyper-autophagy state and restoring it to 'normal' levels [17].
Digging into the supplementary data within the publication, the authors state that one of the mechanisms of action in improved mitophagy is likely due to the decrease in phosphorylated s6, a downstream marker of mTOR activity [17].
In the 20 years from 1999-2019, there has been an increased prevalence of Alzheimer's, with >6 million adults aged >60 suffering from the disease. Although there is a genetic component to this disease via the APO-e4 gene, changes to dietary, exercise, and other environmental influences may accelerate its progression.
The ApoE4 gene, also known as the apolipoprotein E4 gene, refers to a specific variant of the apolipoprotein E (APOE) gene. APOE is a gene that provides instructions for producing a protein called apolipoprotein E, which plays a crucial role in lipid metabolism. The APOE gene has three common variants: APOE2, APOE3, and APOE4. The E4 variant is associated with an increased risk of developing neurodegenerative disorders. The APOE4 gene has been identified as a major genetic risk factor for late-onset Alzheimer's disease.
Since 2019, Professor Matt Kaeberlein has been calling for clinical trials into Rapamycin as a therapeutic tool to prevent and slow Alzheimer's disease. He even wrote an article published in Science Translation Medicine titled "Rapamycin and Alzheimer's disease: Time for a clinical trial?" [19].
This drug has a compelling pre-clinical record in mice showing outstanding benefits, including reducing amyloid-β(Aβ) deposition, reducing pathogenic tau phosphorylation and abundance of misfolded tau species, including neurofibrillary tangles, restoring cerebral blood flow, preserving blood-brain barrier integrity, and improving cognitive function [19].
In a model of Alzheimer's, the potential of the mitochondrial membrane decreases and the permeability increases, which leads to increases in ROS production and cytochrome C release, eventually inducing neuronal apoptosis.
Recent studies have begun to explore the molecular processes by which Rapamycin alters brain metabolism, with mitochondrial function and particular mitophagy and quality control (i.e., the synthesis of new mitochondria and subsequent degradation of old/damaged mitochondria) playing a critical role.
In aged mice, the treatment with Rapamycin increases mitochondrial protein synthesis (i.e., biogenesis & building of new mitochondria). Utilizing state-of-the-art stable isotope tracer techniques, Professor Benjamin Miller's research group at Oklahoma Medical Research Foundation has observed no effect of Rapamycin on brain mitochondrial protein synthesis in younger mice [20].
However, in an older cohort, Rapamycin induced a nearly 40% increase in mitochondrial protein synthesis, with no distinct effect on mTOR, AMPK, or protein aggregates (i.e., insoluble amyloid-beta plaques (Aβ) and neurofibrillary tangles of tau protein) [20].
Stable isotope tracer techniques are powerful tools used in scientific research, specifically in the field of protein synthesis. These techniques involve the use of stable isotopes, which are non-radioactive forms of elements (typically carbon-13 (13C), nitrogen-15 (15N), and deuterium (2H), to track and measure the metabolic processes related to protein synthesis in living organisms.
On the other side of mitochondrial quality control, others have sought to determine how Rapamycin can force the removal of damaged/dysfunctional mitochondria within the aging brain. Although no research group to date has used the powerful Mito-QC model to study mitophagy dynamics, several authors have probed the effects of Rapamycin on molecular markers associated with mitophagy in models of Alzheimer's [21].
An 8-week study of daily Rapamycin (1mg/kg/day) enhanced learning and memory, synaptic plasticity, and the expression of synapse-related proteins [21]. Although not as sensitive as the Mito-QC model, the authors attempted to determine how autophagy proteins co-localize in an attempt to quantify mitophagy.
In this method, they chose to assess the co-localization of TOM20 (an outer mitochondrial membrane protein) & LC3B (a key marker of the autophagosome), in which greater co-localization translates to increased mitochondrial-specific mitophagy. Rapamycin treatment increased mitophagy by >400% compared to the non-treatment group, which was concurrent with an increase in the protein expression of P62, Parkin and LC3B within the mitochondrial fraction of the brain. This is a crucial step to isolate the mitochondria during analysis, as the whole-brain tissue sampled in this study actually observed a decline in mitophagy-related markers [21].
Nicotinamide Riboside (NR) and Nicotinamide Mononucleotide (NMN)
Nicotinamide Riboside (NR) and Nicotinamide Mononucleotide (NMN) are precursors to NAD+ (Nicotinamide Adenine Dinucleotide), a coenzyme critical for cellular energy metabolism. NAD+ serves as an essential helper molecule, enabling the conversion of nutrients into ATP, the primary energy currency of the cell. Beyond energy production, NAD+ plays a pivotal role in activating sirtuins, a family of proteins associated with cellular repair, longevity, mitochondrial function, and mitophagy.
While Urolithin A (UA) shares the goal of improving mitochondrial health, it operates via a distinct mechanism. Rather than relying on NAD+ pathways and sirtuin activation, UA directly enhances mitophagy—the targeted clearance of damaged mitochondria. This direct action on mitochondrial quality control allows UA to bypass the dependency on NAD+ elevation, making it a unique and complementary intervention for promoting mitochondrial health.
The mechanistic differences between NR/NMN and UA open the possibility for synergistic benefits. NR and NMN improve mitochondrial function by replenishing NAD+ levels and activating downstream pathways like sirtuins, which influence mitochondrial biogenesis and overall energy metabolism. Meanwhile, UA works by clearing out dysfunctional mitochondria, ensuring that only healthy and efficient mitochondria remain. Together, these compounds could address multiple aspects of mitochondrial quality control—NAD+ boosting for energy production and sirtuin activation, paired with UA’s mitophagy-driven cleanup—potentially amplifying their overall effects on cellular health and longevity.
Spermidine
Spermidine is a naturally occurring polyamine that induces autophagy, including mitophagy, by modulating multiple cellular signaling pathways. It is recognized for its potential anti-aging effects and its potential to improve both cellular and mitochondrial function.
Although both Spermidine and Urolithin A promote autophagy, Urolithin A stands out for its specific targeting of mitophagy. This focus on mitochondrial quality control may make Urolithin A particularly effective in addressing diseases or conditions where mitochondrial dysfunction is a key factor, such as neurodegenerative diseases or metabolic disorders. While beneficial, Spermidine's broader action on autophagy may not offer the same level of precision in targeting mitochondrial health specifically.
Urolithin A stands out among longevity protocols due to its targeted promotion of mitophagy, which is the selective clearance of damaged mitochondria without strongly inhibiting mTOR. This distinguishes it from other compounds like Rapamycin, which broadly induce autophagy and mitophagy through significant mTOR inhibition, potentially leading to side effects. Urolithin A's reliance on AMPK activation and its direct enhancement of mitophagy also set it apart from NAD+-boosting compounds like NR and NMN, which work through more generalized pathways that impact multiple cellular processes. [4, 5]
While other compounds, such as Spermidine, also promote both autophagy and mitophagy, Urolithin A's unique, mitophagy-specific action makes it particularly promising for therapies targeting mitochondrial dysfunction, which is a hallmark of aging and many age-related diseases, such as neurodegenerative disorders, metabolic diseases, and cardiovascular conditions.
Urolithin A and Inflammation
Mitochondrial dysfunction, marked by a decline in mitochondrial efficiency, is a key factor in aging and is closely associated with chronic inflammation and a wide range of age-related diseases. Mitochondria generate energy for the cell through oxidative phosphorylation, a process that naturally produces reactive oxygen species (ROS) as a byproduct. While moderate levels of ROS are essential for cellular signaling and immune defense, excessive ROS can damage mitochondrial DNA, proteins, and lipids, compromising mitochondrial function.
Over time, ROS-induced damage accumulates, creating a vicious cycle. Damaged mitochondria produce even more ROS, further exacerbating cellular stress and inflammation. This cycle not only accelerates mitochondrial dysfunction but also drives chronic inflammation, a hallmark of aging that underlies many degenerative conditions, including metabolic disorders, neurodegenerative diseases, cardiovascular diseases, and muscle weakness.
The body has natural antioxidant defenses to counteract oxidative stress, including enzymes like manganese-dependent superoxide dismutase (MnSOD), which neutralizes ROS to protect mitochondrial integrity. However, with age, these defenses weaken, resulting in greater oxidative damage and unresolved inflammation. Chronic, low-grade inflammation—often referred to as "inflammaging"—is a major contributor to the decline of tissues and organs over time.
Urolithin A (UA) addresses mitochondrial dysfunction by enhancing mitophagy and facilitating the selective removal of damaged mitochondria. This improvement in mitochondrial quality directly reduces excessive ROS production, thereby interrupting the harmful feedback loop that perpetuates oxidative stress and inflammation.
By restoring efficient mitophagy, UA mitigates the inflammatory responses that result from mitochondrial dysfunction. Damaged mitochondria, which often emit pro-inflammatory signals, are replaced by fully functional mitochondria capable of meeting cellular energy demands without generating excess ROS. Research indicates that UA’s ability to enhance mitochondrial quality has systemic benefits, including the reduction of oxidative stress and inflammation. [4, 5]
UA’s capacity to control inflammation through improved mitochondrial health has profound implications for age-related conditions. Chronic inflammation fueled by mitochondrial dysfunction is a central feature of diseases such as osteoarthritis, sarcopenia (age-related muscle loss), and neurodegenerative disorders. Therefore, by targeting the root cause—damaged mitochondria—UA offers a potential therapeutic strategy to prevent or mitigate these conditions, promoting healthier aging.
Urolithin A, Mitophagy, and Muscle Health
As we mentioned in our analysis of the human study by Singh et al. (2022), Urolithin A (UA) supplementation significantly improved muscle strength and aerobic endurance in middle-aged adults. Key performance indicators such as peak oxygen consumption (VO₂) and the 6-minute walk test (6MWT) demonstrated measurable gains. These improvements were linked to an increase in proteins associated with mitophagy and mitochondrial metabolism in skeletal muscle, highlighting UA’s ability to enhance mitochondrial health [4].
With age, declining mitophagy efficiency leads to the accumulation of dysfunctional mitochondria, which produce less ATP and higher levels of reactive oxygen species (ROS). This contributes to oxidative stress, inflammation, and reduced muscle performance. Given mitochondria's critical role in ATP production for muscle cells, maintaining their function is essential for muscle strength and endurance [4].
UA addresses this issue by supporting the removal of dysfunctional mitochondria and fostering the regeneration of healthy ones. By improving mitochondrial quality control, UA enhances energy production, reduces oxidative stress, and sustains muscle function.
In a related study by Liu et al. (2022), titled Effect of Urolithin A Supplementation on Muscle Endurance and Mitochondrial Health in Older Adults, researchers explored Urolithin A's effects on muscle health in elderly individuals aged 65 to 90. Participants received either 1000 mg of Urolithin A or a placebo for four months. The study measured muscle endurance, improvements in 6-minute walk distance, and biomarkers tied to mitochondrial and cellular health. The specific muscles tested for endurance were the tibialis anterior (TA) in the leg and the first dorsal interosseus (FDI) in the hand, both of which are critical for mobility and grip strength. [6]
The findings showed that participants receiving Urolithin A experienced significant improvements in muscle endurance compared to those in the placebo group. Muscle endurance was assessed by counting the number of muscle contractions before fatigue set in. Those given Urolithin A could perform more contractions, demonstrating better muscle endurance. This effect can be attributed to the enhanced mitochondrial function, as healthy mitochondria produce more ATP, reducing the energy deficit that leads to muscle fatigue. [6]
Additionally, Urolithin A reduced plasma levels of biomarkers such as acylcarnitines, ceramides, and C-reactive protein (CRP). Elevated levels of these molecules are often associated with poor mitochondrial function and increased inflammation. Acylcarnitines are byproducts of fatty acid metabolism, and their elevated presence indicates mitochondrial inefficiency, as the mitochondria cannot properly use fatty acids for energy. These acylcarnitine levels are an indication that the mitochondria work more efficiently to produce energy. [6]
The reduction in CRP, a marker of systemic inflammation, suggests that Urolithin A also mitigates the inflammatory processes contributing to muscle fatigue and degradation. Chronic inflammation is known to impair muscle function by disrupting cellular processes and promoting oxidative stress, so reducing CRP levels reflects a decrease in these harmful effects. [6]
The study also revealed that participants receiving Urolithin A walked further in the 6-minute walk test—an average of 60.8 meters compared to 42.5 meters for the placebo group. This improvement in walking distance indicates enhanced muscle endurance and physical performance, further emphasizing Urolithin A's role in improving mitochondrial function. [6]
Urolithin A's Neuroprotective Effects
While Urolithin A is often recognized for its ability to improve muscle performance, emerging research also suggests it offers neuroprotective benefits through its capacity to enhance mitophagy. This process is critical in tissues like the brain and auditory cells, where mitochondrial dysfunction contributes to age-related diseases, including hearing loss.
A notable study by Cho et al. (2022), titled UA attenuates auditory cell senescence by activating mitophagy, explored whether UA could protect auditory cells from aging-related decline by improving mitochondrial function. Hearing loss is often linked to aging, as the mitochondria in auditory cells—which convert sound into electrical signals for the brain—become less efficient over time. This decline in mitochondrial efficiency leads to oxidative stress, where reactive oxygen species (ROS), harmful byproducts of cellular metabolism, accumulate and damage cells. Excess ROS causes mitochondrial damage, disrupting the cell's energy production and leading to cell death, a major contributor to hearing loss as we age. [7]
The study used a model in which HEI-OC1 auditory cells and cochlear explants (tissue samples from the inner ear) were exposed to hydrogen peroxide (H₂O₂) to mimic the oxidative stress that occurs during aging. Hydrogen peroxide generates high levels of ROS, which damage the cells' proteins, DNA, and mitochondria, accelerating the aging process. After inducing this oxidative stress, the researchers treated the cells with Urolithin A to test whether it could counteract the damage. [7]
The results were striking. The cells exposed to H₂O₂ showed significant increases in p53 and p21, two key markers of cellular aging. These proteins control the cell cycle and promote senescence (a state in which cells stop dividing and lose function). At the same time, the markers that promote mitophagy were reduced, suggesting that the cells' ability to clear out damaged mitochondria was impaired. [7]
However, after treatment with Urolithin A, the levels of p53 and p21 significantly decreased, indicating that the aging process was being reversed. Additionally, there was a marked increase in proteins associated with mitophagy and mitochondrial metabolism, meaning Urolithin A had reactivated the cell's ability to clean out dysfunctional mitochondria. This is crucial because when damaged mitochondria accumulate, they produce higher ROS, perpetuating the cycle of cellular damage and aging. [7]
The findings from Cho et al. (2022) strongly suggest that Urolithin A holds promise as a neuroprotective agent, particularly in combating age-related mitochondrial dysfunction in auditory cells. By enhancing mitophagy, UA helps remove damaged mitochondria, reduce oxidative stress, and break the vicious cycle of mitochondrial damage and cellular aging. The study demonstrated that UA reverses cellular aging markers and restores mitochondrial function, leading to improved energy production and a decrease in harmful ROS accumulation. These protective effects of UA could have significant implications for preventing or slowing neurodegenerative conditions linked to mitochondrial dysfunction. [7]
Urolithin A, Mitophagy, and Metabolic Health
In addition to its well-documented benefits for muscle and neuroprotective health, Urolithin A has been shown to improve insulin sensitivity, which is essential for regulating blood sugar levels and preventing insulin resistance. Insulin sensitivity refers to how effectively the body's cells respond to insulin, the hormone that helps glucose enter cells for energy. When insulin sensitivity is high, cells efficiently absorb glucose, reducing the risk of conditions like type 2 diabetes (T2D). When sensitivity declines, the body needs more insulin to manage blood sugar levels, which can eventually lead to insulin resistance and metabolic disorders. [8]
UA has also demonstrated potential in addressing various metabolic conditions, including obesity and metabolic syndrome. These conditions are often linked to excessive fat accumulation, chronic inflammation, and poor mitochondrial function. By promoting healthier fat metabolism and reducing inflammation, UA may help mitigate the risk factors associated with these disorders. [8]
Toney et al. (2019) conducted a critical study, titled UA, a Gut Metabolite, Improves Insulin Sensitivity Through Augmentation of Mitochondrial Function and Biogenesis, investigating UA's role in improving metabolic health, specifically its effects on obesity and insulin resistance. In this study, male mice were fed a high-fat diet for 12 weeks and given either UA or a control solution (dimethyl sulfoxide). The researchers measured how well the mice responded to insulin by tracking blood sugar levels after administering both glucose and insulin. Additionally, the study looked at the impact of UA on fat accumulation in the liver, the size of fat cells, mitochondrial DNA synthesis (critical for energy production), and the expression of inflammatory markers. [8]
The results showed that the mice treated with UA exhibited improved insulin sensitivity, meaning their bodies were more efficient at using insulin to lower blood sugar levels. UA also reduces fat buildup in the liver, a key indicator of metabolic dysfunction often seen in obesity. Furthermore, the size of fat cells decreased, which is beneficial because larger fat cells tend to release more inflammatory molecules, contributing to chronic inflammation and insulin resistance. [8]
Beyond fat metabolism, UA had a profound impact on mitochondrial function. Mitochondria are the energy-producing centers of cells, and mitochondrial health is crucial for maintaining metabolic efficiency. The study found that UA increased the production of mitochondrial DNA in the liver, indicating enhanced mitochondrial biogenesis (the creation of new mitochondria), which is essential for improving energy production and overall metabolic health. [8]
Another important finding of the study was UA's effect on the immune system, particularly its regulation of macrophages—a type of white blood cell involved in both inflammation and tissue repair. The study revealed that UA shifted macrophage activity from the pro-inflammatory M1 type to the anti-inflammatory M2 type. M1 macrophages are known to promote inflammation, which is harmful in metabolic diseases, while M2 macrophages help resolve inflammation and support tissue healing. This shift suggests that UA improves insulin sensitivity and fat metabolism and reduces chronic inflammation, which is often a key driver of metabolic disorders. [8]
Urolithin A's Cardioprotective Effects
Urolithin A (UA) has shown potential benefits for cardiovascular health, particularly by enhancing mitophagy in heart cells. By replacing dysfunctional mitochondria with healthy, functional ones, UA improves energy production and the contractile capacity of heart muscle cells, allowing the heart to pump blood more efficiently. Additionally, by removing damaged mitochondria, UA reduces oxidative stress, which can otherwise damage blood vessels and contribute to heart disease.
A study by Nishimoto et al. (2023) investigated UA's effects on vascular endothelial function (VEF) in human subjects. VEF refers to the health of the endothelium, the thin layer of cells lining blood vessels that play a critical role in regulating blood flow, pressure, and the passage of nutrients and waste. Impaired VEF is often linked to atherosclerosis—the buildup of plaque in arteries—and increased inflammation, both of which are major risk factors for heart disease.
In this randomized clinical trial, participants with poor VEF and limited ability to process UA from ellagic acid were given either a placebo or, 10 mg, or 50 mg of UA daily for 12 weeks. Flow-mediated dilation (FMD), a test to assess how well blood vessels expand in response to increased blood flow, was used to evaluate VEF. Reduced FMD is indicative of poor cardiovascular health. [9]
The study found that participants who took 50 mg of UA showed a significant increase in gut bacteria diversity, a positive marker of gut health. There was also a trend toward improved FMD scores, suggesting enhanced vascular function. Additionally, the study observed an increase in certain beneficial gut bacteria—Rumini Clostridium 5, (Eubacterium) coprostanoligenes, and (Eubacterium) hallii—in those taking UA. These bacteria are associated with lower blood cholesterol levels, improved insulin sensitivity, and better circadian rhythm regulation, all contributing to cardiovascular health. [9]
The study suggests that Urolithin A enhances vascular endothelial function and improves gut health. This suggests a potential link between gut microbiome changes and cardiovascular improvements, highlighting the importance of focusing on gut health as a novel approach to promoting heart health.
Side Effects and Safety
Urolithin A (UA) has demonstrated significant health benefits, primarily by enhancing mitophagy, which is crucial for improving mitochondrial function, energy production, and overall cellular health. Research shows that UA can extend both lifespan and healthspan by supporting muscle strength, neuroprotection, and metabolic health, including improved insulin sensitivity. Its positive effects on cardiovascular health further highlight its potential to enhance vascular function and reduce the risk of heart disease.
UA has been extensively studied for safety across various clinical trials and is generally considered safe within recommended dosage ranges. The US FDA has classified UA as "Generally Recognized as Safe" (GRAS) for daily doses between 250 mg and 1,000 mg. In a landmark human clinical trial by Andreux et al. (2019), no significant adverse effects were observed, even with prolonged administration over several weeks. The trial involved healthy, sedentary elderly individuals who received either single or multiple doses of UA ranging from 500 to 1,000 mg over a 4-week period. UA was detected in the blood at all doses, and higher doses led to beneficial changes, including a reduction in acylcarnitines—molecules that transport fat into mitochondria for energy production. This decrease suggests that UA helps the body use fat more efficiently, with no significant side effects reported. [10]
Despite its strong safety profile, there is variability in how individuals respond to UA, mainly due to differences in gut microbiota, which are responsible for converting dietary precursors into the compound. Some individuals may experience mild digestive discomfort, such as bloating, gas, or nausea. In rare cases, allergic reactions like itching or rash may occur. This variability highlights the importance of personalized dosing and consulting with healthcare providers, particularly for individuals with existing health conditions or those taking medications. [10]
While short-term studies have not identified major safety concerns, the long-term effects of UA remain an area for further research. Animal studies have explored the effects of high doses (up to 3451 mg/kg in males and 3826 mg/kg in females) over 90 days, with no observed toxicity or adverse changes in blood chemistry or organ function. However, these findings have not yet been confirmed in humans. As such, long-term supplementation should be approached with caution, especially for populations like pregnant or breastfeeding women, where safety data is limited. [11]
While UA shows significant promise as a dietary supplement, particularly for aging and muscle health, there are areas that warrant further investigation. Future research should focus on the effects of higher doses exceeding 1,000 mg and longer-term use in human populations. In the next section, we will explore these research gaps and ongoing studies that aim to uncover more about the benefits and mechanisms of UA. [11]
Future Directions and Research
While Urolithin A (UA) research shows great promise, several gaps still need to be addressed. One major area is the impact of UA on humans with different demographic profiles. The effectiveness of UA largely depends on how it is processed by gut microbiota, which varies significantly across populations due to diet, lifestyle, genetics, and environment. For instance, in the Nishimoto et al. (2023) study, participants with improved vascular endothelial function (VEF) after UA intake had a specific gut microbiota profile characterized by a low Bacillota/Bacteroidota ratio. These findings suggest that more research is needed to explore how different microbiota compositions affect UA's efficacy. [12]
Another challenge is the need for a more detailed understanding of how UA promotes mitophagy at the molecular level. Measuring mitophagy directly in humans is difficult. Current research relies on analyzing molecular indicators like the levels of proteins associated with mitophagy, such as PINK1, Parkin, and LC3B. These provide insights, but the extent to which mitophagy occurs needs to be confirmed. Further trials should combine UA administration with advanced tools to better measure mitophagy.
For example, Ginefra et al. (2024) used an innovative tool called MitoQC to assess mitophagy in mice. This tool uses fluorescent proteins to track changes in mitochondria during mitophagy. Their study showed increased T cells with high mitophagy activity in mice given UA, supporting its role as a mitophagy inducer. Future human studies using similar tools will help validate these findings. [13]
UA holds potential beyond current applications. Its role in removing dysfunctional mitochondria makes it especially useful in fields like exercise physiology, where it could aid in recovery by reducing muscle damage and inflammation. There's also growing interest in its potential for cancer therapy, as studies like Ginefra et al. (2024) have shown that UA boosts FOXO1. This protein enhances the immune system's ability to fight tumors. Future research could explore UA's use with other compounds like Rapamycin, NR, and Resveratrol and how individual genetic differences influence UA's effectiveness, paving the way for personalized medicine. [13]
Conclusion
Urolithin A (UA) holds significant potential in promoting healthy aging by enhancing mitophagy, the process of clearing out damaged mitochondria and improving mitochondrial function. By focusing on mitochondrial health, UA supports muscle performance, metabolic health, neuroprotection, and cardiovascular function. Its ability to reduce inflammation and oxidative stress further highlights its potential in combating a wide range of age-related diseases.
While UA has a strong safety profile with minimal short-term side effects, more research is needed to explore its long-term effects, particularly across diverse populations. Additionally, its relationship with gut microbiota and the precise molecular mechanisms involved require further investigation.
As ongoing studies continue to explore UA's benefits in fields such as exercise recovery, cancer therapy, and personalized medicine, the compound shows great promise for expanding its applications in clinical practice and improving the lifespan and health span of many.
- Hodzic Kuerec, A., Lim, X. K., Khoo, A. L. Y., Sandalova, E., Guan, L., Feng, L., & Maier, A. B. (2024). Targeting aging with urolithin A in humans: A systematic review. Ageing Research Reviews, 85, 102406. https://doi.org/10.1016/j.arr.2024.102406
- Luan, P., D'Amico, D., Andreux, P. A., Laurila, P. P., Wohlwend, M., Li, H., Imamura de Lima, T., Place, N., Rinsch, C., Zanou, N., & Auwerx, J. (2021). UA improves muscle function by inducing mitophagy in muscular dystrophy. Science translational medicine, 13(588), eabb0319. https://doi.org/10.1126/scitranslmed.abb0319
- Huang, J. R., Zhang, M. H., Chen, Y. J., Sun, Y. L., Gao, Z. M., Li, Z. J., Zhang, G. P., Qin, Y., Dai, X. Y., Yu, X. Y., & Wu, X. Q. (2023). UA ameliorates obesity-induced metabolic cardiomyopathy in mice via mitophagy activation. Acta pharmacologica Sinica, 44(2), 321–331. https://doi.org/10.1038/s41401-022-00919-1
- Singh, A., D'Amico, D., Andreux, P. A., Fouassier, A. M., Blanco-Bose, W., Evans, M., Aebischer, P., Auwerx, J., & Rinsch, C. (2022). UA improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell reports. Medicine, 3(5), 100633. https://doi.org/10.1016/j.xcrm.2022.100633
- D'Amico, D., Olmer, M., Fouassier, A. M., Valdés, P., Andreux, P. A., Rinsch, C., & Lotz, M. (2022). UA improves mitochondrial health, reduces cartilage degeneration, and alleviates pain in osteoarthritis. Aging cell, 21(8), e13662. https://doi.org/10.1111/acel.13662
- Liu, S., D'Amico, D., Shankland, E., Bhayana, S., Garcia, J. M., Aebischer, P., Rinsch, C., Singh, A., & Marcinek, D. J. (2022). Effect of UA Supplementation on Muscle Endurance and Mitochondrial Health in Older Adults: A Randomized Clinical Trial. JAMA network open, 5(1), e2144279. https://doi.org/10.1001/jamanetworkopen.2021.44279
- Cho, S. I., Jo, E. R., & Song, H. (2022). UA attenuates auditory cell senescence by activating mitophagy. Scientific reports, 12(1), 7704. https://doi.org/10.1038/s41598-022-11894-2
- Toney, A. M., Fan, R., Xian, Y., Chaidez, V., Ramer-Tait, A. E., & Chung, S. (2019). UA, a Gut Metabolite, Improves Insulin Sensitivity Through Augmentation of Mitochondrial Function and Biogenesis. Obesity (Silver Spring, Md.), 27(4), 612–620. https://doi.org/10.1002/oby.22404
- Nishimoto, Y., Fujisawa, K., Ukawa, Y., Kudoh, M., Funahashi, K., Kishimoto, Y., & Fukuda, S. (2023). Effect of UA on the improvement of vascular endothelial function depends on the gut microbiota. Frontiers in nutrition, 9, 1077534. https://doi.org/10.3389/fnut.2022.1077534
- Andreux, P. A., Blanco-Bose, W., Ryu, D., Burdet, F., Ibberson, M., Aebischer, P., Auwerx, J., Singh, A., & Rinsch, C. (2019). The mitophagy activator UA is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nature metabolism, 1(6), 595–603. https://doi.org/10.1038/s42255-019-0073-4
- Heilman, J., Andreux, P., Tran, N., Rinsch, C., & Blanco-Bose, W. (2017). Safety assessment of UA, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association, 108(Pt A), 289–297. https://doi.org/10.1016/j.fct.2017.07.050
- Ginefra, P., Hope, H. C., Chiang, Y. H., Nutten, S., Blum, S., Coukos, G., & Vannini, N. (2024). Urolithin-A Promotes CD8+ T Cell-mediated Cancer Immunosurveillance via FOXO1 Activation. Cancer research communications, 4(5), 1189–1198. https://doi.org/10.1158/2767-9764.CRC-24-0022
- Marshall, R. N. (2022, June 11). Healthspan Research Review: Clearing out the cellular trash: The importance of mitophagy in the removal of damaged & dysfunctional mitochondria. Healthspan. https://gethealthspan.com/science/article/mitophagy-removal-damaged-dysfunctional-mitochondria
- Plummer JD, Johnson JE. Extension of Cellular Lifespan by Methionine Restriction Involves Alterations in Central Carbon Metabolism and Is Mitophagy-Dependent. Front Cell Dev Biol 2019; 7. doi:10.3389/FCELL.2019.00301/PDF.
- Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. doi:10.1038/nature14300.
- Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 2016; 22: 1428–1438.
- Mito T, Vincent AE, Faitg J, Taylor RW, Khan NA, Mcwilliams TG et al. Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metab 2022; 34. doi:10.1016/j.cmet.2021.12.017.
- Ubaida-Mohien C, Spendiff S, Lyashkov A, Moaddel R, Macmillan NJ, Filion ME et al. Unbiased Proteomics, Histochemistry, and Mitochondrial DNA Copy Number Reveal Better Mitochondrial Health in Muscle of High Functioning Octogenarians. Elife 2022; 11. doi:10.7554/ELIFE.74335.
- Kaeberlein M, Galvan V. Rapamycin and Alzheimer's disease: Time for a clinical trial? Sci Transl Med 2019; 11. doi:10.1126/SCITRANSLMED.AAR4289.
- Reid JJ, Linden MA, Peelor FF, Miller RA, Hamilton KL, Miller BF. Brain Protein Synthesis Rates in the UM-HET3 Mouse Following Treatment With Rapamycin or Rapamycin With Metformin. J Gerontol A Biol Sci Med Sci 2020; 75: 40.
- Wang H, Fu J, Xu X, Yang Z, Zhang T. Rapamycin Activates Mitophagy and Alleviates Cognitive and Synaptic Plasticity Deficits in a Mouse Model of Alzheimer's Disease. The Journals of Gerontology: Series A 2021; 76: 1707–1713.