Mitophagy is essentially mitochondrial-specific autophagy.
Measuring mitophagy in humans is currently impossible, but mouse models allow us to measure this process in incredible detail.
Exercise induces large increases in mitophagy process but returns to baseline levels ~24hrs post-exercise. Therefore, repeated bouts of exercise, either daily or as many times per week, are essential to maintain healthy mitochondria and remove the damaged & dysfunctional ones.
Nutritional supplements, in particular Urolithin A, can increase mitophagy, and improve mitochondrial health independent of exercise. However, a direct study of exercise + Urolithin A/Placebo is likely needed to determine its true effect on mitophagy compared to exercise.
Rapamycin has been shown to induce autophagy via decreased mTOR activity. New research is showing this also has a beneficial effect on mitophagy and can have a beneficial effect on brain mitochondria via the synthesis and biogenesis of new mitochondria, as well as the clearance of damaged & dysfunction mitochondria through mitophagy. Overall, this may treat, offset, or at least slow down the progression of neurodegenerative diseases such as Alzheimer's.
Overview
The use of the word 'autophagy' is becoming ever-present in the health, longevity, and anti-aging community. However, the role of mitochondrial-specific autophagy is relatively less understood, termed 'mitophagy.' This process is almost identical to traditional autophagy; however, it focuses solely on targeting damaged, dysfunctional, and ineffective mitochondria for their removal and degradation.
Recent evidence has begun to show how critical this process is in maintaining proteostasis within a cell or tissue and how dysregulation of this process can result in the accumulation of damaged mitochondria, leading to rapid disease progression (Parkinson's, Alzheimer's, T2D, Liver Disease & Muscular Dystrophy).
Here we will discuss some of the exciting recent advances in our understanding of this process and, more importantly, how we can potentially manipulate this with exercise, diet, and pharmacological therapies in an effort to improve cellular health, metabolic health, and longevity.
What is mitophagy?
Mitophagy is a selective process by which damaged or dysfunctional mitochondria are selectively targeted for degradation within cells and is an essential mechanism for maintaining mitochondrial quality control and overall cellular homeostasis [1]. This process involves the engulfment of damaged mitochondria by specialized vesicles called autophagosomes, which then fuse with lysosomes, leading to the degradation and recycling of the targeted mitochondria's contents [1].
Molecular Regulation of Mitophagy & How We Measure This Process
In humans, the process of mitophagy is impossible to directly measure and is limited primarily to snapshots of particular molecular readouts of genes/proteins associated with mitophagy [2]. For example, tissue samples (typically muscle biopsies) from human volunteers, following various interventions, can be analyzed to determine the gene expression or protein abundance of molecular targets such as Pink1, Parkin, P62, LC3B, ULK1 and FUNDC1 [3].
Another side of the mitophagy equation is the molecular machinery that controls mitochondrial dynamics. This is effectively the molecular control of mitochondrial fusion (i.e., the merging of smaller mitochondria to form larger, tubular mitochondria) and mitochondrial fission (i.e., the removal of damaged fragments of mitochondrial to undergo degradation) [4]. These processes are controlled by several proteins, but most notably, OPA-1 [5] & MFN-2 [6], and DRP-1 [7] & FIS-1 [8], which control fusion and fission, respectively.
Unfortunately, these markers, typically assessed via western blot or PCR, only give a snapshot of the time the tissue sample was obtained and do not directly infer mitophagy is occurring. Nevertheless, these indirect and semi-quantitative measures of indirect mitophagy still give us an insight into how this process may be regulated.
A more sophisticated model is the MitoQC (mitochondrial quality control) model developed by Dr Tom McWilliams (now at the University of Helsinki, Finland) and Professor Ian Ganley at The University of Dundee's Protein Phosphorylation Unit [9].
The MitoQC model involves using a fluorescent reporter protein (green fluorescent protein and mCherry) targeted to the outer mitochondrial membrane via the protein FIS-1 [9]. These reporter proteins are designed to accumulate on the mitochondrial membrane under normal conditions within a pH of ~7.2 [10].
However, when mitophagy is induced, the damaged or dysfunctional mitochondria are targeted for degradation via the acidic environment within the lysosome (~pH 4.8), leading to the clearance of the pH-sensitive green fluorescent protein from the mitochondria, resulting in the red puncta of mCherry displaying mitophagy [10].
By monitoring the clearance of the fluorescent reporter protein via a sophisticated confocal microscope and live imaging techniques, researchers can assess the occurrence and efficiency of mitophagy within the cells and tissues at time points or in real time over several hours.
This technique allows for highly sensitive quantitative analysis of mitophagy dynamics. It provides insights into the molecular mechanisms involved in mitochondrial quality control and has been instrumental in advancing our understanding of mitophagy and its regulation.
Can We Manipulate Mitophagy?
Mitochondrial dysfunction has been implicated as one of the nine hallmarks of aging, along with other hallmarks such as epigenetic alterations, genomic instability, cellular senescence, stell cell exhaustion, telomere attrition, dysregulated nutrient sensing, loss of proteostasis and altered intracellular communication [11].
Mitochondrial dysfunction can be subcategorized into decreased mitochondrial biogenesis, decreased respiratory capacity, increased ROS production, a decline in mitochondrial content, and impairments in mitochondrial quality control and removal of damaged/dysfunctional mitochondria via mitophagy [12].
Emerging evidence suggests that enhancing mitophagy can positively impact longevity and lifespan. Several studies involving genetic mutation, dietary manipulations, or pharmacological interventions have demonstrated that promoting mitophagy extends the lifespan in various model organisms, including yeast [13], worms [14], flies, and mice [15].
Moreover, impaired mitophagy has been linked to age-related diseases, such as neurodegenerative disorders [16] and cardiovascular disease [17]. Dysfunction in mitophagy pathways can lead to the accumulation of damaged mitochondria, oxidative stress, and inflammation, contributing to the pathogenesis of these diseases.
Enhancing mitophagy through pharmacological interventions or lifestyle modifications holds promise for mitigating age-related diseases and promoting healthy aging. Here we will briefly discuss how exercise, nutrition, and pharmacological interventions can improve mitophagic regulation, improve mitochondrial quality control as we age, and prevent the accumulation of damaged mitochondria within cells & tissues.
Exercise and Mitophagy
The effect of exercise, particularly endurance/HIIT exercise, has significant evidence regarding its impact on mitochondrial biogenesis and mitochondrial respiration, resulting in improved health [18] and increased exercise performance [19].
Until recently, the role of exercise & mitophagy was relatively unclear. In 2017 and 2020, two post-doctoral researchers in the laboratory of Professor Zhen Yan at the University of Virginia's Centre for Skeletal Muscle Research published incredible work displaying the role of exercise on muscle mitophagy.
The first, Dr. Rhianna Laker (now a Research Scientist & Team Leader at AstraZeneca), showed (in mice) that 90 minutes of treadmill running induced a 45% increase in mitophagy (i.e., green:red ratio) [20].
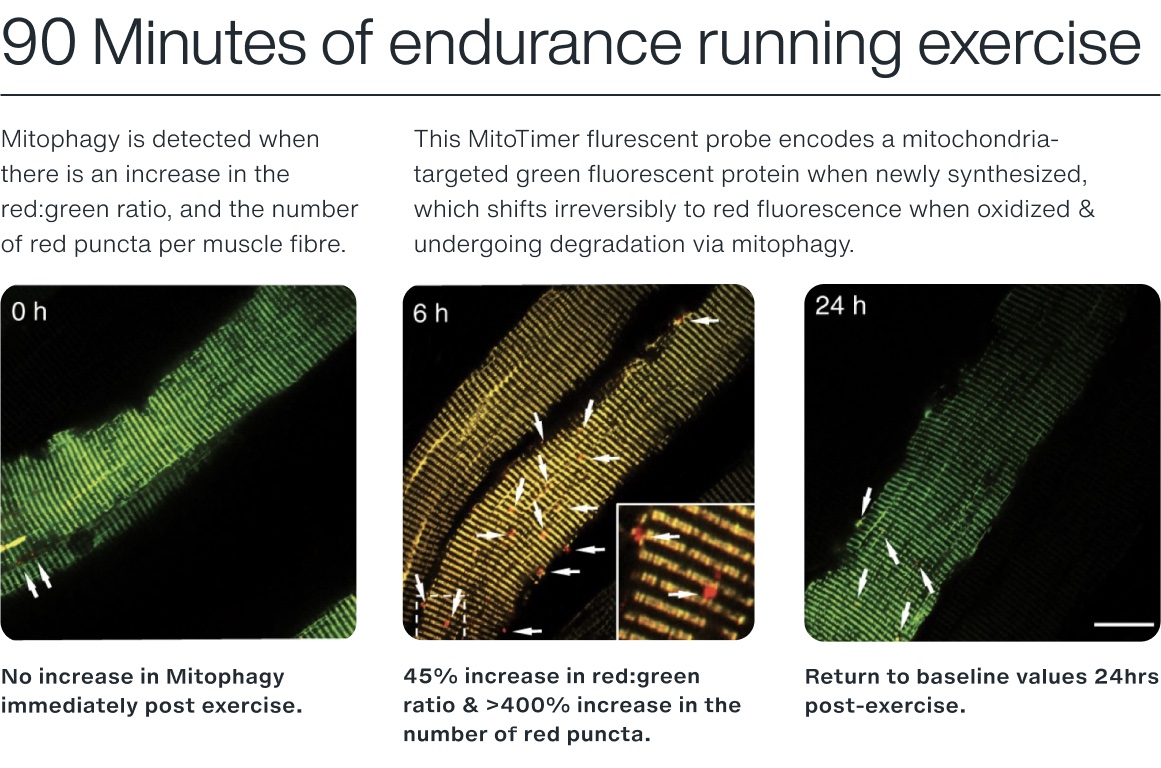
Using a similar reporter model (MitoTimer) [21], we can see how exercise results in the fluorescent color change 6hrs post-exercise. Notably, there was a >400% increase in the number of pure red fluorescent puncta in the fibers, indicating significantly elevated levels of mitochondria being degraded via the lysosome [20].
In this study, the group displayed that mitophagy is preceded by the phosphorylation of AMPK and its downstream target, ULK-1. To confirm AMPK's role in mitophagy, the authors used a mouse model to knock out the dominant α2 subunit of AMPK and subjected the mice to the same exercise. Following 90 minutes of treadmill exercise, no mitophagy was detected [20].
Secondly, a follow-up to this by Dr Joshua Drake (now an assistant professor at Virginia Tech) showed the crucial role of AMPK in mitophagy [22]. For the first time, he and the team showed that AMPK is actually localized to the mitochondrial membrane following exercise or energetic stress.
Within this study, the team tested the hypothesis of metformin, a method of inducing energetic stress and thereby increasing mitophagy. Metformin directly impaired complex I, as previously reported, with little to no effect on mitochondrial respiration [22]. However, as previously reported with exercise, metformin increased mitochondrial localized AMPK [22], indicating the potential for metformin to induce skeletal muscle mitophagy independent of exercise.
Nutrition and Mitophagy
The world-leading laboratory of Professor Johan Auwerx at the École Polytechnique Fédérale in Lausanne, Switzerland, have collaborated with Amazentis (a Nestle Health Science company) on the development and now commercialization of a nutritional supplement aimed at improving mitophagy.
This collaboration has earned the team several high-profile publications in Nature Metabolism, Nature Medicine, and JAMA. The translation research process from cells to c.elegans (worms), mice and even humans have consistently observed improvements in mitochondrial health and, notably, increased mitophagy.
What is interesting about natural Urolithin A is how it is synthesized. The process is incredibly variable and only occurs in approximately 40% of the human population [23], hence why supplementation is essential to elevate blood concentration [24]. Being a 'UA responder requires the appropriate gut microbiome, and in particular, bacteria species, Gordonibacter urolithinfaciens and Ellagibacter isourolithinifaciens, which convert the ellagitannins from pomegranates and other sources into active Urolithin A [24].
Initial evidence in 2016 from Professor Auwerx's lab and the start of the Amazentis Urolithin A research showed this nutritional supplement increased lifespan in c.elegans by 45%, which was primarily due to improved mitochondrial function [25].
To explore this further, the authors determine with a sensitive reporter model of mitophagy (termed 'MitoKeima' – A similar model to MitoQC). Following a 24hr exposure to Urolithin A in muscle cells, mitophagy was increased by ~104% compared to control [25]. In mice [25], middle-aged adults [26], and older adults [27], Urolithin A has been observed to increase the gene & protein expression of proteins related to mitophagy.
Data from randomized control trials of middle-aged adults supplementing 1,000mg/day for 4 months reported improvements in muscle strength (~12%) and endurance performance (~12%). Muscle biopsies obtained pre & post-intervention from the vastus lateralis muscle of the quadriceps showed a significant increase in genes related to mitophagy, TCA cycle, and fatty acid oxidation, overall indicating improvements in mitochondrial health & function [26].
Pharmacological-Induced Mitophagy
It's well established that Rapamycin induces autophagy by suppressing mTOR activity. Therefore, it should likely have an effect on mitochondrial-specific autophagy, right?
Surprisingly, there's not too much evidence on the effects of Rapamycin on mitophagy. However, a recent 2022 paper published in Cell Metabolism by Dr Tom McWilliams has characterized the role of Rapamycin in mitophagy in a model of mitochondrial disease using the previously mentioned reporter models, which he developed during his PhD [28]. This model has some similarities to aging, in which older adults, especially octogenarians >80yrs age, display severe mitochondrial abnormalities [29].
The study shines light on the role of this powerful drug on mitochondrial quality control. The model of mitochondrial disease typically displays respiratory chain defects, 'ragged-red fibers' (expressing pathogenic mitochondria DNA variants & severe accumulation of damaged mitochondria) [28].
This study showed that the treatment of Rapamycin at a dose of 8mg/kg/day for 70 days resulted in a 125% increase in mitophagy [28]. The authors speculated that in aging and mitochondrial disease, there's hyperactivation of autophagy-mediated degradation as indicated by the accumulation of P62 and phosphorylated ubiquitin in muscle fibres [28]. Notably, compared to healthy control patient samples, patients with mitochondrial disease displayed an >80-fold increase in the autophagy adapter protein P62, with rapamycin treatment causing a ~90% decrease in the hyper-autophagy state and restoring it to 'normal' levels [28].
Digging into the supplementary data within the publication, the authors state that one of the mechanisms of action in improved mitophagy is likely due to the decrease in phosphorylated s6, a downstream marker of mTOR activity [28].
In the 20 years from 1999-2019, there has been an increased prevalence of Alzheimer's, with >6 million adults aged >60 suffering from the disease. Although there is a genetic component to this disease via the APO-e4 gene, changes to dietary, exercise, and other environmental influences may accelerate its progression.
Since 2019, Professor Matt Kaeberlein has been calling for clinical trials into Rapamycin as a therapeutic tool to prevent and slow Alzheimer's disease. He even wrote an article published in Science Translation Medicine titled "Rapamycin and Alzheimer's disease: Time for a clinical trial?" [30].
This drug has a compelling pre-clinical record in mice showing outstanding benefits, including reducing amyloid-β(Aβ) deposition, reducing pathogenic tau phosphorylation and abundance of misfolded tau species, including neurofibrillary tangles, restoring cerebral blood flow, preserving blood-brain barrier integrity, and improving cognitive function [30].
In a model of Alzheimer's, the potential of the mitochondrial membrane decreases and the permeability increases, which leads to increases in ROS production and cytochrome C release, eventually inducing neuronal apoptosis.
Recent studies have begun to explore the molecular processes by which Rapamycin alters brain metabolism, with mitochondrial function and particular mitophagy and quality control (i.e., the synthesis of new mitochondria and subsequent degradation of old/damaged mitochondria) playing a critical role.
In aged mice, the treatment with Rapamycin increases mitochondrial protein synthesis (i.e., biogenesis & building of new mitochondria). Utilizing state-of-the-art stable isotope tracer techniques, Professor Benjamin Miller's research group at Oklahoma Medical Research Foundation has observed no effect of Rapamycin on brain mitochondrial protein synthesis in younger mice 31.
However, in an older cohort, Rapamycin induced a nearly 40% increase in mitochondrial protein synthesis, with no distinct effect on mTOR, AMPK, or protein aggregates (i.e., insoluble amyloid-beta plaques (Aβ) and neurofibrillary tangles of tau protein) [31].
On the other side of mitochondrial quality control, others have sought to determine how Rapamycin can force the removal of damaged/dysfunctional mitochondria within the aging brain. Although no research group to date has used the powerful Mito-QC model to study mitophagy dynamics, several authors have probed the effects of Rapamycin on molecular markers associated with mitophagy in models of Alzheimer's [32].
An 8-week study of daily Rapamycin (1mg/kg/day) enhanced learning and memory, synaptic plasticity, and the expression of synapse-related proteins [32]. Although not as sensitive as the Mito-QC model, the authors attempted to determine how autophagy proteins co-localize in an attempt to quantify mitophagy.
In this method, they chose to assess the co-localization of TOM20 (an outer mitochondrial membrane protein) & LC3B (a key marker of the autophagosome), in which greater co-localization translates to increased mitochondrial-specific mitophagy. Rapamycin treatment increased mitophagy by >400% compared to the non-treatment group, which was concurrent with an increase in the protein expression of P62, Parkin and LC3B within the mitochondrial fraction of the brain. This is a crucial step to isolate the mitochondria during analysis, as the whole-brain tissue sampled in this study actually observed a decline in mitophagy-related markers [32].
What does this all mean?
Imagine your body's cells as a bustling city, with each cell serving as a home to a multitude of mitochondria. Over time, some of these mitochondria become damaged or dysfunctional, much like waste accumulating in a city. But fear not, for your body has a team of diligent garbage men called mitophagy, ready to clear out the cellular trash.
Just like garbage collectors roam the streets, mitophagy patrols the inner workings of your cells, identifying and targeting those dysfunctional mitochondria for removal. They possess specialized tools and mechanisms to distinguish the good from the bad, much like how trained eyes can distinguish recyclables from general waste.
When a mitochondrion becomes damaged, it displays molecular signals. These signals act as a beacon, attracting the attention of the mitophagy crew. Once identified, the garbage men surround the damaged mitochondrion, enclosing it in a specialized membrane, similar to placing a hazardous waste bag around a bin.
With precision and efficiency, the mitophagy team then proceeds to engulf the damaged mitochondrion within a double-membrane structure, resembling a garbage truck devouring its contents. This structure, called an autophagosome, safely engulfs the malfunctioning mitochondrion, isolating it from the healthy ones.
Just like a garbage truck transporting its load to a waste processing facility, the autophagosome transports the captured mitochondrion to a cellular recycling center called the lysosome. The lysosome acts as a cellular incinerator, breaking down the damaged mitochondrion into its component parts, recycling valuable materials, and disposing of waste products.
In this process, mitophagy acts as the tireless garbage men, diligently patrolling your cells, identifying, and removing damaged mitochondria to maintain a clean and functional cellular environment.
By eliminating dysfunctional mitochondria, mitophagy helps prevent the accumulation of cellular waste, reducing the risk of oxidative damage and maintaining overall cellular health, much like efficient waste management contributes to a cleaner and healthier city.
Just as we rely on the dedication of garbage collectors to keep our cities clean and liveable, our bodies depend on the vigilant work of mitophagy to ensure the efficient removal of damaged mitochondria and maintain cellular well-being. So, next time you see a garbage truck passing by, remember the incredible parallel of mitophagy, tirelessly working to keep your cells free from the waste of dysfunctional mitochondria.
- Youle RJ, Narendra DP. Mechanisms of mitophagy. 2011. doi:10.1038/nrm3028.
- Hamacher-Brady A, Brady NR. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cellular and Molecular Life Sciences 2016; 73: 775–795.
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20: 31–42.
- Liu YJ, Mcintyre RL, Janssens GE, Houtkooper RH. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. 2020. doi:10.1016/j.mad.2020.111212.
- Tezze C, Romanello V, Desbats MA, Salviati L, Scorrano L, Correspondence MS. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab 2017; 25: 1374-1389.e6.
- Bell MB, Bush Z, McGinnis GR, Rowe GC. Adult skeletal muscle deletion of Mitofusin 1 and 2 impedes exercise performance and training capacity. J Appl Physiol 2019; 126: 341–353.
- Favaro G, Romanello V, Varanita T, Andrea Desbats M, Morbidoni V, Tezze C et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun 2019; 10: 1–17.
- Losó n OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 2013; 24: 659–667.
- McWilliams TG, Prescott AR, Allen GFG, Tamjar J, Munson MJ, Thomson C et al. Mito-QC illuminates mitophagy and mitochondrial architecture in vivo. Journal of Cell Biology 2016; 214: 333–345.
- Mcwilliams TG, Ganley IG. Chapter 41 Investigating Mitophagy and Mitochondrial Morphology In Vivo Using mito-QC: A Comprehensive Guide. doi:10.1007/978-1-4939-8873-0_41.
- Schmauck-Medina T, Molière A, Lautrup S, Zhang J, Chlopicki S, Madsen HB et al. New hallmarks of ageing: a 2022 Copenhagen ageing meeting summary. 2022.www.aging-us.com (accessed 2 Jun2023).
- Sun N, Youle RJ, Finkel T. Molecular Cell The Mitochondrial Basis of Aging. doi:10.1016/j.molcel.2016.01.028.
- Plummer JD, Johnson JE. Extension of Cellular Lifespan by Methionine Restriction Involves Alterations in Central Carbon Metabolism and Is Mitophagy-Dependent. Front Cell Dev Biol 2019; 7. doi:10.3389/FCELL.2019.00301/PDF.
- Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. doi:10.1038/nature14300.
- Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 2016; 22: 1428–1438.
- Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA et al. Mitophagy and Alzheimer's Disease: Cellular and Molecular Mechanisms. Trends Neurosci 2017; 40: 151–166.
- Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol 2016; 594: 509–525.
- Ringholm S, Gudiksen A, Frey Halling J, Qoqaj A, Meizner Rasmussen P, Prats C et al. Impact of Aging and Lifelong Exercise Training on Mitochondrial Function and Network Connectivity in Human Skeletal Muscle. The Journals of Gerontology: Series A 2022; 2022: 1–11.
- Granata C, Oliveira RSF, Little JP, Renner K, Bishop DJ. Mitochondrial adaptations to high-volume exercise training are rapidly reversed after a reduction in training volume in human skeletal muscle. FASEB Journal 2016; 30: 3413–3423.
- Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 2017; 8: 1–13.
- Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH et al. A novel mitotimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. Journal of Biological Chemistry 2014; 289: 12005–12015.
- Drake JC, Wilson RJ, Laker RC, Guan Y, Spaulding HR, Nichenko AS et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc Natl Acad Sci U S A 2021; 118: e2025932118.
- Amico DD', Andreux PA, Valdés P, Singh A, Rinsch C, Auwerx J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. 2021. doi:10.1016/j.molmed.2021.04.009.
- Singh A, Davide D’amico •, Pénélope •, Andreux A, Dunngalvin G, Kern • Timo et al. Direct supplementation with Urolithin A overcomes limitations of dietary exposure and gut microbiome variability in healthy adults to achieve consistent levels across the population. Eur J Clin Nutr 2022; 76: 297–308.
- Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nature Medicine 2016 22:8 2016; 22: 879–888.
- Singh A, Amico DD’, Andreux LA, Aebischer P, Auwerx J, Rinsch C et al. Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. doi:10.1016/j.xcrm.2022.100633.
- Liu S, D’Amico D, Shankland E, Bhayana S, Garcia JM, Aebischer P et al. Effect of Urolithin A Supplementation on Muscle Endurance and Mitochondrial Health in Older Adults: A Randomized Clinical Trial. JAMA Netw Open 2022; 5: e2144279–e2144279.
- Mito T, Vincent AE, Faitg J, Taylor RW, Khan NA, Mcwilliams TG et al. Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metab 2022; 34. doi:10.1016/j.cmet.2021.12.017.
- Ubaida-Mohien C, Spendiff S, Lyashkov A, Moaddel R, Macmillan NJ, Filion ME et al. Unbiased Proteomics, Histochemistry, and Mitochondrial DNA Copy Number Reveal Better Mitochondrial Health in Muscle of High Functioning Octogenarians. Elife 2022; 11. doi:10.7554/ELIFE.74335.
- Kaeberlein M, Galvan V. Rapamycin and Alzheimer's disease: Time for a clinical trial? Sci Transl Med 2019; 11. doi:10.1126/SCITRANSLMED.AAR4289.
- Reid JJ, Linden MA, Peelor FF, Miller RA, Hamilton KL, Miller BF. Brain Protein Synthesis Rates in the UM-HET3 Mouse Following Treatment With Rapamycin or Rapamycin With Metformin. J Gerontol A Biol Sci Med Sci 2020; 75: 40.
- Wang H, Fu J, Xu X, Yang Z, Zhang T. Rapamycin Activates Mitophagy and Alleviates Cognitive and Synaptic Plasticity Deficits in a Mouse Model of Alzheimer's Disease. The Journals of Gerontology: Series A 2021; 76: 1707–1713.
Related studies