From Cholesterol to ApoB and Lp(a): The Particle Revolution in Heart Health
ApoB as a Superior Marker of Cardiovascular Risk: Apolipoprotein B (apoB) provides a clearer picture of cardiovascular risk than traditional cholesterol metrics. Because every atherogenic lipoprotein particle (VLDL, IDL, LDL, and Lp(a)) carries one molecule of apoB, its concentration directly reflects the number of circulating particles driving atherosclerosis. Unlike LDL-C, which measures cholesterol content, apoB quantifies the true burden of atherogenic exposure—making it a more powerful and consistent predictor of cardiovascular events.
How Metabolic Dysfunction Accelerates Atherogenesis: Chronic hyperinsulinemia and insulin resistance fuel atherogenesis by increasing hepatic VLDL output, which elevates apoB particle numbers. At the same time, metabolic dysfunction damages the endothelium through oxidative stress, inflammation, and reduced nitric oxide availability, all of which promote vascular stiffness and plaque formation. These mechanisms link metabolic health directly to the intensity of atherogenic particle damage.
The Role of Arterial Wall Retention and Inflammation: ApoB-containing particles become trapped in the arterial wall when they bind to glycosaminoglycans (GAGs), setting off immune activation and chronic inflammation. Glycation—common in insulin-resistant states—further increases this binding, intensifying plaque buildup. Individual differences in immune response determine how aggressively inflammation progresses, helping explain why some people develop severe atherosclerosis despite moderate lipid elevations.
Evidence Linking ApoB Reduction to Lower Disease Risk: Across genetic and clinical studies, lowering apoB particle counts consistently reduces cardiovascular risk. In a cohort of 654,000 individuals, identical apoB reductions—regardless of mechanism—led to equivalent decreases in coronary disease. Trials like IMPROVE-IT and FOURIER confirm that smaller apoB reductions translate to measurable event declines, though benefits taper at very low levels. Notably, natural antibodies against apoB-100 correlate with a 45% lower risk of heart attack, underscoring its central role in disease progression.
Lipoprotein(a): From Evolutionary Advantage to Modern Risk: Lipoprotein(a), or Lp(a), once aided wound repair but now contributes to inflammation and clot formation, raising cardiovascular risk 2–4× and markedly increasing rates of aortic stenosis. Roughly 20% of people have genetically elevated Lp(a), which diet or statins cannot modify. PCSK9 inhibitors can lower it by 20–30%, while antisense oligonucleotide therapies may cut levels by up to 90%, offering the most promising path for targeted intervention.
Setting Safe and Clinically Meaningful ApoB Targets: Optimal apoB levels appear to mirror those seen in healthy children—around 30–40 mg/dL—while most guidelines recommend keeping levels below 70 mg/dL for high-risk individuals. Further lowering may bring diminishing returns, suggesting a personalized balance between aggressive reduction and biological necessity. The future of prevention lies in targeting apoB precisely, not just suppressing cholesterol broadly.
Introduction
In this research review, we explore the evolving understanding of cardiovascular health, underscored by a significant paradigm shift from a traditional cholesterol-centric perspective to a nuanced appreciation of lipoprotein behavior, particularly focusing on the roles of apoB and Lp(a) in cardiovascular disease.
This shift was catalyzed by Dr. Allan Sniderman's influential narrative review in JAMA Cardiology, which introduced the ApoB Particle Model of Atherogenesis, challenging the traditional cholesterol models and underscoring the superior predictive power of apoB particle counts for cardiovascular risk.
We begin by examining the conditions under which cholesterol becomes harmful, emphasizing how metabolic dysfunctions— notably elevated insulin levels and mitochondrial dysfunction—drive the progression of atherosclerosis. This discussion sets the stage for a deeper exploration of the pivotal roles and transport mechanisms of lipoproteins, essential for a comprehensive understanding of cardiovascular risk.
Particular emphasis is placed on the density of these lipoproteins and its influence on the levels of atherogenic apoB-containing lipoproteins. We further explore the clinical implications of this model, examining how reductions in apoB particles significantly decrease the risk of heart disease and the underlying biological mechanisms that support this relationship.
Additionally, we analyze the risks associated with lipoprotein(a) or Lp(a)—a lesser known but particularly harmful lipoprotein. We discuss Lp(a)'s evolutionary roles, its transformation into a modern health risk, and strategies for mitigating its impact on cardiovascular health.
We conclude with the difficulties and practical strategies for managing levels of Lp(a) and apoB, this review not only expands our understanding of cardiovascular risk factors but also provides practical guidance on mitigating these risks through lifestyle changes and therapeutic interventions. By bridging the gap between cutting-edge research and clinical practice, this review aims to empower healthcare professionals and patients in their ongoing fight against cardiovascular disease.
When Does Cholesterol Become Dangerous?
Before we discuss the specific role of lipoproteins and their potential to increase the level of cardiovascular disease, let’s first build a foundation of knowledge of the cholesterol-centric view of cardiovascular disease. As we’ll discuss throughout this research review, there is a discordance between the levels of cholesterol and lipoprotein particles that is very important to understanding cardiovascular risk. However, it is important to lay a foundation for understanding how these particles can become harmful.
In the cholesterol-centric view of cardiovascular disease, understanding when and how cholesterol becomes harmful is crucial for preventing cardiovascular diseases. Low-density lipoprotein (LDL) particles are central to this discussion. The danger posed by LDL particles primarily arises when they are retained in the subendothelial space of arterial walls, undergoing oxidation and triggering an inflammatory response. Here’s a detailed breakdown of this process:
Entry and Retention
LDL particles enter the subendothelial space, located just beneath the inner lining of the artery (the endothelium). While most LDL particles typically exit this space without causing harm, problems arise when they become retained. Factors like increased blood LDL levels and changes in the structure of the arterial wall can enhance this retention. Once trapped, these particles are prone to undergo harmful modifications, such as oxidation [1].
Oxidation
Oxidation refers to the chemical modification of LDL particles through reactions with free radicals or other oxidants present in the arterial wall. This modification significantly changes the nature of LDL, turning it into an agent that actively contributes to arterial inflammation and damage. Oxidized LDL is not recognized by normal cellular receptors and instead attracts immune cells that attempt to clear these modified particles from the arterial walls [2].
Inflammatory Response
The immune response to oxidized LDL is a double-edged sword. While it aims to clear these harmful particles, it often exacerbates the problem. Macrophages, a type of white blood cell, absorb the oxidized LDL, transforming into foam cells. These foam cells are a hallmark of early atherosclerotic plaques. The accumulation of foam cells and the secretion of inflammatory substances by these cells further damage the arterial wall and attract more immune cells, creating a vicious cycle of inflammation [3].
The Final Stage: Atherosclerosis Development
As this inflammatory process continues, atherosclerotic plaques begin to form and grow. These plaques consist of lipids, inflammatory cells, smooth muscle cells, and connective tissue. Over time, these plaques can become large enough to significantly narrow the arteries and harden, a condition known as arteriosclerosis. This narrowing can impair blood flow and increase the risk of acute cardiovascular events like heart attacks and strokes if a plaque ruptures, leading to the formation of a clot [4].
Therefore, while the size of LDL particles and their number can influence their likelihood of becoming harmful, the key factors that drive their atherogenic potential are their retention in the arterial wall, subsequent oxidation, and the inflammatory response they trigger. This comprehensive understanding of the atherogenic process underscores the importance of targeting these mechanisms to prevent and treat atherosclerosis.
The Interrelated Nature of Insulin Levels, ApoB, Triglycerides, and Atherosclerotic Cardiovascular Disease (ASCVD)
Now we have provided a basis for how cholesterol becomes harmful. Let’s now consider how metabolic health factors affect this process. The interplay between insulin levels specifically, apoB, triglycerides, and heart disease is critical to understanding overall cardiovascular health risk and the levers we have to control that risk.
The Role of Insulin in Cardiovascular Disease
Insulin is a central regulatory hormone in both glucose and lipid metabolism. Normally, it facilitates the uptake of glucose from the bloodstream into cells, particularly muscle and fat cells, allowing them to use glucose for energy. Insulin also plays a role in fat metabolism by inhibiting the breakdown (lipolysis) of fat in adipose (fat) tissue. This process prevents the excessive release of fatty acids into the bloodstream [5].
When insulin resistance develops, cells in the body do not respond effectively to normal levels of insulin. This resistance is particularly pronounced in muscle, liver, and fat cells, the primary targets of insulin action. To compensate for reduced efficacy, the pancreas produces more insulin, leading to hyperinsulinemia. This syndrome is associated with an increased risk of obesity, type 2 diabetes, and cardiovascular diseases [6].
Insulin’s Impact on Lipids
Insulin resistance significantly impacts lipid levels, particularly triglycerides and apoB-containing lipoproteins.
Triglycerides
Normally, insulin helps regulate triglyceride levels by controlling their synthesis in the liver and inhibiting the breakdown of fat in adipose tissue. However, in states of insulin resistance and compensatory hyperinsulinemia, the liver's production of triglycerides increases.
Meanwhile, the reduced efficacy of insulin means that the inhibition of fat breakdown is not as effective, leading to an overall increase in triglycerides in the bloodstream. High triglyceride levels are a well-known risk factor for cardiovascular diseases because they contribute to the development of atherosclerosis by fostering the formation of atherogenic lipoproteins [7].
ApoB and LDL
Insulin resistance stimulates the liver to produce more very-low-density lipoprotein (VLDL), which is notably rich in triglycerides. VLDL particles undergo metabolic processes in the bloodstream that gradually convert them into low-density lipoprotein (LDL), the primary carriers of cholesterol in the blood.
This increase in VLDL production escalates the levels of apoB, the main protein component of VLDL and LDL particles. ApoB is a critical indicator of cardiovascular risk because each apoB-containing lipoprotein has the potential to contribute to plaque formation in the arteries [8].
In our discussion of ApoB, we’ll go into more detail about how these lipoproteins contribute to heart disease. For now, just note that ApoB-containing lipoproteins are particularly atherogenic. ApoB particles have a higher propensity to penetrate the arterial wall, where they can become trapped and oxidized, initiating an inflammatory response that leads to the development of atherosclerotic plaques.
Hyperinsulinemia and Endothelial Dysfunction
Not only do elevated levels change your lipid profile, it can wreak havoc on the endothelial cells lining our blood vessels, leading to endothelial dysfunction—a precursor to atherosclerosis.
Oxidative Stress and Reduced Nitric Oxide
Insulin at high concentrations can stimulate endothelial cells to produce more reactive oxygen species (ROS). These ROS are chemically reactive molecules containing oxygen, which, in excess, lead to oxidative stress—a state where the oxidative damage outweighs the body's ability to detoxify the reactive intermediates or repair the resulting damage.
The oxidative stress diminishes the bioavailability of nitric oxide (NO), a critical molecule that endothelial cells produce to regulate vascular relaxation and maintain the flexibility of blood vessels. Nitric oxide also inhibits platelet aggregation and adhesion, which are crucial factors in preventing thrombosis. When NO levels decrease, the risk of hypertension, thrombosis, and atherosclerosis increases significantly [9, 10].
Inflammation and Endothelial Activation
Elevated insulin levels also enhance the release of pro-inflammatory cytokines like IL-6 and TNF-α. These cytokines are key players in the inflammatory response.
The cytokines activate endothelial cells, increasing their expression of adhesion molecules like VCAM-1 and ICAM-1. These molecules play a vital role in the recruitment and attachment of immune cells, such as monocytes and T-cells, to the endothelium. This interaction is a critical step in the development of endothelial inflammation and atheroma formation, laying the groundwork for plaque development and progression in atherosclerosis [11, 12].
Insulin and Endothelial Cell Death
Chronic hyperinsulinemia contributes to endothelial dysfunction not only through mechanisms like oxidative stress and inflammation but also by directly inducing cell death in endothelial cells.
Persistent high insulin levels can lead to oxidative stress, which in turn activates pro-apoptotic signaling pathways within endothelial cells. These pathways include the activation of stress-related kinases and caspases, which are enzymes that play essential roles in the process of apoptosis, or programmed cell death.
The loss of endothelial cells through apoptosis compromises the integrity and function of the endothelial layer. This disruption can expose the underlying cellular matrix and vascular smooth muscle to circulating lipids and inflammatory cells, accelerating the process of atherosclerosis and contributing to the formation of unstable plaques, which are more prone to rupture and can lead to heart attacks or strokes [13].
Insulin Increases Vascular Stiffness
Elevated insulin levels stimulate the production of endothelin-1, a potent vasoconstrictor that increases vascular resistance and blood pressure. Increased production of endothelin-1 can exacerbate hypertension and further impair endothelial function by promoting vascular stiffness and reducing arterial compliance. This, in turn, heightens the risk of cardiovascular events such as heart attacks and strokes due to impaired blood flow and increased arterial pressure [14].
Tools to Combat Hyperinsulinemia and Endothelial Dysfunction
It’s clear that part of the strategy to reduce our risk of cardiovascular disease involves addressing hyperinsulinemia. We have very effective (and cheap) tools at our disposal for this.
Diet and Exercise
Most of the tools we have to optimize our healthspan are free. Adopting a diet low in refined carbohydrates and sugars can significantly improve insulin sensitivity. Such dietary changes reduce the glycemic load, lessening the demand on the pancreas to produce insulin. Regular physical activity also enhances muscle uptake of glucose and improves insulin sensitivity at the cellular level, which can help stabilize insulin levels and reduce its impact on the endothelium [15].
Antioxidants and Anti-inflammatory Agents (Diet and Supplementation)
Antioxidants and anti-inflammatory agents can play a role in alleviating oxidative stress and inflammation, two major contributors to endothelial dysfunction. These substances help neutralize reactive oxygen species and reduce the inflammatory response within the vascular system, thereby protecting the endothelial cells from further damage and improving vascular function [16].
Insulin Sensitizers (Medications)
Drugs such as metformin and thiazolidinediones (e.g., pioglitazone) are used to improve insulin sensitivity. These medications enhance the ability of cells to respond to insulin, thereby reducing the need for the pancreas to produce excessive amounts. By lowering insulin levels, these drugs help mitigate the endothelial damage caused by hyperinsulinemia and can indirectly reduce the risk of cardiovascular disease by improving overall metabolic health [17].
Take Home Point: Endothelial dysfunction is an early marker of atherosclerosis and increases the risk of cardiovascular events such as heart attacks and strokes. Addressing hyperinsulinemia through lifestyle changes and medication can significantly impact cardiovascular health.
Lipids 101: Understanding Lipoproteins and Their Role in Lipid Transport
We have established the role of cholesterol and metabolic health in changing our relative risk of heart disease. Now we are going to go deeper. It turns out that cholesterol itself is not a great predictor of occurrence. More specifically, it is the lipoproteins that carry it around that cause the harm.
To understand the role of lipoproteins in increasing the occurrence of heart disease, let’s first do a Lipids 101 tutorial.
Lipids and Their Solubility Challenge
Lipids, including fats, oils, and cholesterol, are essential molecules in biology, but their hydrophobic nature presents a significant transportation challenge in the bloodstream. Because lipids do not dissolve in water, they cannot freely travel in the blood plasma, which consists mainly of water (about 90%). The analogy of trying to mix olive oil with water illustrates this challenge well. When mixed, oil and water separate, with the oil forming a distinct layer on top. This separation occurs because lipids are nonpolar and do not interact well with water, a polar solvent.
Lipoproteins: The Submarines of Lipid Transport
To overcome this challenge, lipids are transported in the blood via lipoproteins, which are specially structured particles that can navigate the watery environment of the bloodstream effectively. Think of them as specialized delivery trucks carrying essential lipid cargo through the aqueous plasma. These "trucks" are meticulously designed to navigate the circulatory system efficiently.
Lipoproteins are spherical particles with a core composed of triglycerides (TGs) and cholesteryl esters (CEs). Triglycerides are the main form of stored energy in the body, and cholesteryl esters are a storage form of cholesterol. This core is surrounded by a monolayer of phospholipids and free cholesterol.
Phospholipids, which have both hydrophilic (water-attracting) and hydrophobic (water-repelling) properties, form the outer layer of the lipoprotein particle. The hydrophilic "head" of the phospholipid is oriented outward to interact with the aqueous environment, while the hydrophobic "tail" faces inward, interacting with the lipids inside the lipoprotein. This arrangement allows the lipoproteins to remain soluble in the blood plasma while safely encasing the hydrophobic lipids within [18].
The Role of Apolipoproteins
These structures function like submarines or delivery trucks, encapsulating lipids to transport them to various parts of the body where they are needed. The lipoproteins travel through the bloodstream, delivering lipids to cells for energy production, hormone synthesis, and other critical biological functions. The design of lipoproteins is crucial for the efficient distribution of lipids, preventing the accumulation of lipids in blood vessels, which could lead to blockages and cardiovascular diseases.
The spherical design of lipoproteins is key to their function. This shape allows for a uniform distribution of the hydrophilic (water-attracting) heads of phospholipids and free cholesterol on the surface, facing the aqueous plasma, while the hydrophobic (water-repelling) tails face inward, keeping the lipid core safe. This structural arrangement enables lipoproteins to efficiently carry hydrophobic lipid molecules through the bloodstream, ensuring that essential lipids reach various tissues and organs where they are needed for vital biological processes [19].
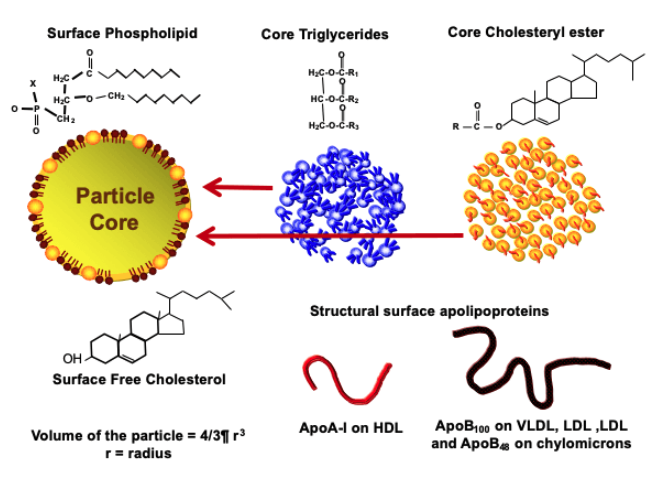
We need this process to work because it is crucial for numerous physiological functions, including energy storage, cell membrane structure, and hormone production. Disruptions in lipoprotein function or lipid transport can lead to metabolic disorders, such as atherosclerosis and cardiovascular diseases [20].
Why the Density of Lipoproteins Really Matters
Many of you are familiar with the different types of cholesterol. LDL and HDL are fairly routine measurements on a lipid panel. However, it’s important to clarify that LDL and HDL are not cholesterol themselves but carriers or transporters of cholesterol and other lipids. This common misnomer oversimplifies their roles and the complex nature of lipid metabolism.
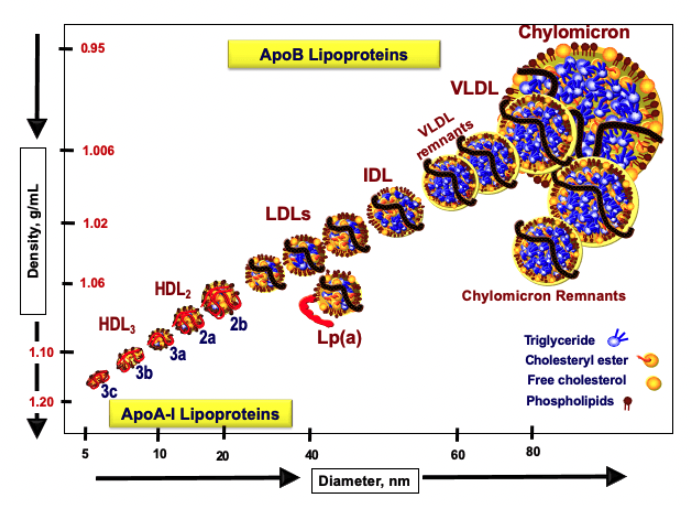
Both LDL and HDL are classes of lipoproteins, distinguished not just by their function but importantly by their density.
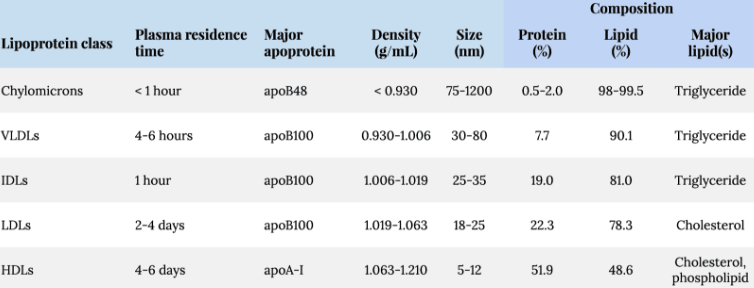
The composition and density of lipoproteins are determined by their lipid-to-protein ratio. Lipoproteins that have a higher proportion of lipids relative to proteins are larger and less dense, making them more buoyant. These include chylomicrons and very-low-density lipoproteins (VLDL). On the other hand, lipoproteins with a higher protein content, such as LDL and HDL, are smaller, denser, and less buoyant.
This structural attribute affects how lipoproteins function within the body. For example, denser lipoproteins can more readily interact with specific receptors on liver cells or become lodged within arterial walls. Understanding these properties is crucial for assessing the risk of lipid-related diseases and the dynamics of lipid metabolism [21].
The classification of lipoproteins by density was facilitated through a technique known as ultracentrifugation. Blood is collected into a sealed tube and subjected to extremely high-speed centrifugation. This process separates blood components based on their density due to the centrifugal force exerted. Lipoproteins, with varying densities, stratify into distinct layers, allowing for their differentiation and study [22].
From this process emerged the classification of lipoproteins into five main categories:
- Chylomicrons (CMs): With the lowest density, these lipoproteins float to the top of the tube. They are primarily produced in the small intestine and are responsible for transporting dietary lipids from the gut to other parts of the body where they can be utilized or stored.
- Very-Low-Density Lipoproteins (VLDLs): Produced in the liver, VLDLs carry triglycerides synthesized from carbohydrates to tissues. They are less dense than other lipoproteins produced by the liver due to their higher lipid content.
- Intermediate-Density Lipoproteins (IDLs): As VLDLs deposit their triglyceride content, they shrink and become denser, forming IDLs. IDLs can further metabolize into LDLs or be taken up by the liver.
- Low-Density Lipoproteins (LDLs): Known colloquially as "bad cholesterol," LDLs are denser than VLDLs and IDLs and are responsible for transporting cholesterol to cells throughout the body. Elevated LDL levels are associated with an increased risk of atherosclerosis.
- High-Density Lipoproteins (HDLs): The densest lipoproteins, HDLs, are involved in reverse cholesterol transport, carrying cholesterol from the body's tissues back to the liver. Known as "good cholesterol," HDLs help to reduce the risk of cardiovascular diseases.
- Lipoprotein(a) [Lp(a)]: Within the LDL category, Lp(a) is a variant that includes an additional protein, apolipoprotein(a). Lp(a) has similar density properties to LDL but poses a unique risk for cardiovascular diseases due to its prothrombotic and atherogenic properties. (We are going to dedicate a whole section to Lp(a)).
Lipid-to-Protein Ratio and Density
The density of these lipoprotein particles is directly related to their composition, specifically their lipid-to-protein ratio.
- High Lipid Content: Particles like CMs and VLDLs have a higher lipid content, making them larger, more buoyant, and therefore less dense. These particles are primarily involved in transporting large amounts of triglycerides and other lipids.
- High Protein Content: Particles such as LDLs and HDLs, which carry fewer lipids relative to their protein content, are smaller and denser. Their compact structure allows them to effectively interact with cellular receptors and participate in crucial metabolic pathways, such as cholesterol recycling and removal.
ApoB and Its Structural Role in Lipoproteins
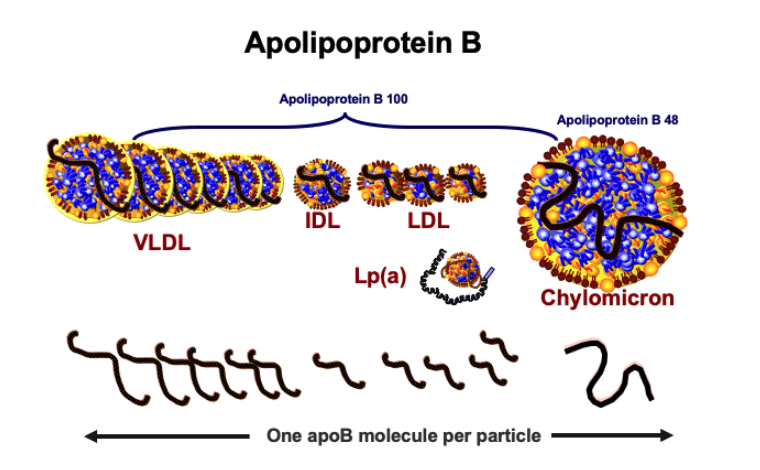
ApoB is a fundamental protein component of several lipoprotein classes involved in lipid transport. ApoB serves as the primary structural protein in chylomicrons (CMs), very-low-density lipoproteins (VLDLs), intermediate-density lipoproteins (IDLs), low-density lipoproteins (LDLs), and lipoprotein(a) [Lp(a)]. It provides structural integrity to these particles and is essential for their proper function in lipid transport.
Each particle of these lipoproteins contains exactly one molecule of apoB. This consistency across different particles makes apoB a valuable marker for estimating the total number of atherogenic lipoprotein particles in the bloodstream [23].
The concentration of apoB in the blood provides a direct measure of the number of lipoprotein particles, which is particularly useful for assessing cardiovascular risk. The total concentration of apoB in the blood is the sum of apoB from each class of lipoproteins carrying it, including CMs, VLDLs, IDLs, LDLs, and Lp(a). This sum gives an accurate count of the total lipoprotein particle number:
Total apoB concentration=CM-apoB+VLDL-apoB+IDL-apoB+LDL-apoB+Lp(a)-apoB
High-density lipoproteins (HDLs) do not contain apoB, highlighting that not all lipoproteins contribute to the total apoB measurement [24].
ApoB as an Indicator of Atherogenic Lipoproteins
ApoB is particularly significant in cardiovascular risk assessment due to its correlation with atherogenic lipoproteins. Since LDL particles, which are known to promote atherosclerosis, comprise over 90% of apoB-containing particles in the plasma, apoB levels predominantly reflect LDL particle concentration. This makes apoB a more precise indicator of atherosclerotic risk compared to traditional measurements of LDL cholesterol.
ApoB levels also account for other atherogenic particles, including Lp(a) and the remnants of VLDLs, both of which can contribute to cardiovascular disease. This comprehensive profiling helps in identifying individuals at higher risk of developing cardiovascular conditions, even when LDL cholesterol levels are not elevated [25].
The measurement of apoB is increasingly recognized in clinical practice as a superior marker for cardiovascular risk, especially in patients with metabolic syndromes or diabetes, where the number of LDL particles may be more critical than their cholesterol content alone.
Why ApoB and Not LDL?
Why should we care more about apoB than just LDL? A recent review in JAMA Cardiology by Allan Sniderman and colleagues highlights a compelling argument: apolipoprotein B (apoB) levels are superior to LDL-C, non-HDL-C, and even LDL particle count (LDL-P) as the best measure of potentially atherogenic lipoproteins [23].
Traditionally, cardiovascular risk assessments have focused primarily on measuring levels of LDL cholesterol (LDL-C), non-HDL cholesterol (non-HDL-C), and LDL particle count (LDL-P). However, these measurements can sometimes provide an incomplete picture of risk:
- LDL-C measures the cholesterol content within LDL particles, but not the number of particles themselves.
- Non-HDL-C includes all atherogenic particles containing cholesterol except for HDL, but it still focuses on cholesterol content rather than particle number.
- LDL-P provides a count of LDL particles but does not account for particles of other lipoproteins that may also contribute to atherosclerosis.
The Superiority of ApoB as a Cardiovascular Risk Marker
ApoB has been recognized as a superior marker because each atherogenic lipoprotein particle contains exactly one apoB molecule, making apoB levels a direct reflection of the number of atherogenic particles. Measuring apoB provides a count of all atherogenic lipoproteins, including LDL, VLDL, IDL, and Lp(a), offering a comprehensive assessment of atherogenic burden.
Sniderman’s study argues that apoB is a better predictor of cardiovascular events than traditional lipid measurements because it reflects the actual number of particles that can penetrate the arterial wall and initiate atherogenesis.
Cholesterol Model vs. ApoB Particle Model of Atherosclerosis
The shift from focusing on cholesterol content to apoB particle numbers offers a more mechanistic insight into how atherosclerosis develops.
The Traditional Cholesterol Model
The traditional cholesterol model posits that the amount of cholesterol within lipoprotein particles (mainly VLDL and LDL) dictates the extent of cholesterol deposition in arteries, leading to atherogenesis.
The apoB Particle Model
In contrast, the apoB model emphasizes that the risk of atherosclerosis depends more critically on the number of apoB-containing particles. These particles are the primary carriers of cholesterol into the arterial wall. Their rate of entrapment in the arterial wall, particularly in the subintimal space, is a key determinant of atherosclerotic risk [26].
Not all apoB particles contribute equally to atherosclerosis. Smaller, cholesterol-depleted apoB particles can penetrate the arterial wall more easily and are more prone to retention than larger, cholesterol-rich particles. Once trapped, smaller particles, although they contain less cholesterol each, can accumulate in greater numbers and potentially release more cholesterol collectively, contributing to greater arterial injury [1]. Larger particles release more cholesterol per particle, but their overall impact may be balanced by the greater number of smaller particles [27].
When it comes to a risk assessment of cardiovascular disease, apoB does not exist in a vacuum. The influence of the arterial wall composition is an important variable in understanding the overall risk of a cardiovascular event.
Influence of Arterial Wall Composition
In addition to apoB burden, the composition and structure of the arterial wall, particularly the glycosaminoglycans (GAGs), play a significant role in the development of atherosclerosis. Arterial wall composition has an important role in the retention of these particles.
GAGs are complex carbohydrates that form part of the extracellular matrix in the arterial wall. They interact with various proteins and lipoproteins, influencing the structural integrity and function of the vasculature. Variations in the structure or composition of GAGs can affect how lipoproteins, particularly apoB-containing particles, are retained within the arterial wall. Different patterns of GAGs might lead to varying degrees of lipoprotein retention, influencing the risk and progression of atherosclerosis.
The process of glycation, where sugar molecules bond non-enzymatically to proteins, can alter the properties of proteins, including apoB.
Glycation of apoB particles can enhance their binding affinity for GAGs, promoting stronger and potentially more pathogenic interactions with the arterial wall. This increased binding could lead to higher retention rates of apoB-containing lipoproteins, contributing to plaque formation and the progression of atherosclerosis. The glycation of apoB and its effects on atherosclerosis development warrant further investigation as a potential therapeutic target or biomarker for cardiovascular risk.
Another variable is individual immune response. The immune response to apoB particles trapped within the arterial wall also varies significantly among individuals, affecting the extent of inflammation and subsequent arterial damage.
When apoB particles are retained in the arterial wall, they can trigger an immune response, leading to the recruitment of immune cells such as macrophages and T-cells. These cells attempt to clear the trapped particles but can also contribute to the inflammatory destruction of the arterial wall. Significant individual differences in how the immune system responds to trapped apoB particles can influence the severity of inflammation and the rate of progression of atherosclerosis [28].
Variations in the structure of glycosaminoglycans and other components of the arterial wall may influence how strongly apoB particles bind and are trapped. Glycation of apoB particles, which promotes their binding, warrants further investigation. Additionally, significant individual variation in the intensity of the immune response to apoB particles trapped within the arterial wall contributes to the inflammatory destruction of the arterial wall [28].
Quantifying Harm of apoB
Despite the significant role of apoB in cardiovascular health, there are currently few treatments specifically designed to reduce apoB levels. However, studies where we can lower apoB levels provide insights into the overall harm of these particles.
Most lipid-lowering therapies, such as statins, primarily target cholesterol synthesis or uptake but do not directly reduce apoB levels. Other therapies might influence apoB indirectly by reducing the production of VLDL in the liver, thereby decreasing the number of apoB-containing particles.
Evidence from studies using autoantibodies targeting apoB provides some data on what lowering apoB can do to reduce heart disease risk.
Studies have shown that some patients develop autoantibodies against specific regions of the apoB protein. These autoantibodies can neutralize the atherogenic potential of apoB-containing particles. A notable study titled, Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis, found that patients with high levels of antibodies targeting the native peptide 210 of apoB-100 had a significantly lower risk of myocardial infarction. This peptide segment of apoB-100 is critical for the particle's interaction with arterial wall components, suggesting that these antibodies may prevent apoB from contributing to plaque formation [29].
The presence of these antibodies was associated with a 45% reduction in the risk of developing a myocardial infarction, underscoring the potential of immunological approaches to manage or prevent CVD [29].
Take Home Point: There was at least a 45% reduction in risk of myocardial infarctions when we reduced the levels of apoB. These immunological studies provide us with some substantiation of the overall risk of elevated levels of apoB particles.
ApoB Particle Number and CHD Risk
The concentration of apoB particles in the bloodstream has been increasingly recognized as a key indicator of CHD risk, perhaps even more so than traditional lipid metrics such as LDL cholesterol (LDL-C) levels.
What Genetic Studies Tell Us
In 2019, cardiologist and genetic epidemiologist Brian Ference from the University of Cambridge and his colleagues published the findings of their study that sought to compare the association of triglyceride-lowering variants in the lipoprotein lipase (LPL) gene and LDL-C-lowering variants in the LDL receptor gene (LDLR) with the risk of cardiovascular disease per unit change in ApoB.
This large-scale study analyzed data from 654,783 individuals to explore the relationship between genetic variants affecting lipid metabolism and CHD risk. The findings highlight that reducing the number of apoB-containing particles, regardless of changes in cholesterol content, effectively lowers the risk of cardiovascular events [30].
The study offers critical insights into the mechanics of lipid-lowering therapies and their effects on cardiovascular health. The research examined the impact of triglyceride-lowering variants in the lipoprotein lipase (LPL) gene and LDL-C–lowering variants in the LDL receptor gene. It found that both types of genetic variants reduce cardiovascular risk comparably when they result in a similar absolute reduction in apoB levels.
The key takeaway is that the absolute change in the number of apoB-containing lipoprotein particles is a more crucial factor for reducing CHD risk than merely altering the cholesterol content within these particles [30].
The findings from this study have profound implications for the treatment of CHD and the management of lipid levels. Traditional approaches that focus solely on lowering LDL-C might not be as effective if they do not also reduce the number of apoB particles. These findings suggest that treatments should aim to decrease the overall number of atherogenic lipoprotein particles to effectively mitigate CHD risk.
The study underscores the need for a shift in focus from merely reducing cholesterol levels to a more comprehensive strategy that includes reducing the concentration of apoB-containing particles. As we'll discuss later in this review, this might involve using therapies that not only alter lipid levels but also decrease the production of lipoproteins at their source, such as statins, PCSK9 inhibitors, or other emerging therapies that impact lipoprotein production and clearance.
Are There Diminishing Returns of Reducing ApoB?
We have some evidence now to support the idea that reducing apoB particles leads to a reduction in the risk of cardiovascular disease. Yet, questions persist about whether the benefits of lowering ApoB continue indefinitely or if they diminish at very low levels.
Evidence for Linear Benefit
The relationship between reducing apoB and decreasing CVD risk has been supported by numerous studies:
IMPROVE-IT Trial: The IMPROVE-IT Trial is a key piece of evidence demonstrating the benefits of intensive lipid-lowering therapy. By combining simvastatin with ezetimibe, the trial showed that further reductions in apoB could lead to additional decreases in cardiovascular events beyond those achieved by statin therapy alone. This suggests a continued benefit in lowering apoB levels for reducing CVD risk [31].
Potential for Diminishing Returns
While the initial reductions in apoB are generally beneficial, the extent of risk reduction may not always remain consistent. Evidence suggests that while significant reductions in apoB can substantially decrease CVD risk, the incremental benefits may taper off at lower concentrations of apoB. This observation has prompted questions about a possible threshold beyond which further reductions might yield minimal additional benefits.
Mendelian randomization, what we saw in the Ference study, provides a method to assess the lifelong impact of genetically determined lower apoB levels. Studies using this approach indicate that individuals with genetically lower apoB levels due to specific variants show a reduced risk of CVD. However, these reductions do not always scale linearly compared to the effects observed with pharmacological interventions, suggesting a potential limit to the benefits of reducing apoB levels.
Clinical Trial Data Involving PCSK9 Inhibitors
Recent trials involving PCSK9 inhibitors, which are highly effective in lowering LDL cholesterol (and by extension apoB), have provided further data.
FOURIER Trial
This trial evaluated the PCSK9 inhibitor evolocumab and found significant reductions in CVD risk. Evolocumab is a monoclonal antibody designed to inhibit proprotein convertase subtilisin-kexin type 9 (PCSK9), a protein that degrades LDL receptors on liver cells, thereby preventing them from removing LDL cholesterol from the blood. This trial sought to assess whether reducing LDL cholesterol levels with evolocumab beyond the levels achieved with statins alone could prevent cardiovascular events.
After 48 weeks, evolocumab reduced LDL cholesterol levels by 59% compared to placebo, lowering median LDL levels from 92 mg per deciliter to 30 mg per deciliter. Treatment with evolocumab significantly reduced the risk of the primary endpoint— of cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization—with 9.8% of patients in the evolocumab group experiencing an event compared to 11.3% in the placebo group [32].
However, the degree of risk reduction began to diminish as LDL cholesterol levels dropped below 30 mg/dL, supporting the concept of diminishing returns at very low apoB levels [32].
The theory behind diminishing returns can be explained through the concept of atherogenic lipoprotein particle burden. At higher apoB levels, more atherogenic particles are available to penetrate the arterial wall, increasing the risk of plaque formation. As apoB levels are reduced, the number of these harmful particles decreases substantially, reducing the likelihood of further arterial damage. At very low apoB levels, the few remaining particles might not significantly contribute to additional risk, thus the marginal benefit of further reductions may become less significant.
Like anything in science, the implications of this data are once again nuanced. While aggressive apoB reduction is beneficial, especially for high-risk patients, it is important to balance this with other aspects of care.
A Closer Look at ApoB Reduction by Analyzing the IMPROVE-IT Trial
The IMPROVE-IT trial is a landmark study that specifically assessed the benefits of adding ezetimibe to statin therapy in patients who had recently experienced an acute coronary syndrome (ACS).
The study involved 18,144 patients who had been hospitalized for ACS within the previous 10 days. These patients were randomized to receive either a combination therapy of simvastatin 40 mg plus ezetimibe 10 mg daily or simvastatin 40 mg plus a placebo.
The goal was to determine whether the addition of ezetimibe to statin therapy, which would further lower LDL cholesterol and consequently apoB levels, could provide additional cardiovascular protection compared to statin therapy alone [31].
The IMPROVE-IT trial provided clear results on the effectiveness of intensive lipid-lowering therapy. Patients in the simvastatin-ezetimibe group achieved a significantly lower mean LDL cholesterol level (53.7 mg/dL) compared to those receiving simvastatin alone (69.5 mg/dL). This reduction is significant as LDL cholesterol carries most of the apoB in the bloodstream, thus a decrease in LDL directly correlates with a decrease in apoB levels.
The primary endpoint of the trial included cardiovascular death, major coronary events, and nonfatal stroke. Over a median follow-up of 6 years, the combination therapy group saw a 6.4% relative risk reduction in these events, with an absolute risk reduction of 2%. This translates to preventing 20 major cardiovascular events per 1,000 patients treated over seven years with the combination therapy.
By demonstrating that lower LDL cholesterol levels lead to improved cardiovascular outcomes, the trial indirectly supports the hypothesis that reductions in apoB particles contribute to reduced cardiovascular risk. This is particularly relevant for high-risk patients, such as those with recent ACS.
The findings bolster the case for intensive lipid-lowering strategies in managing patients post-ACS. They highlight the importance of not only achieving low LDL cholesterol targets but also the broader impact of these targets on lowering apoB and reducing atherogenic particle burden.
What Should Your Target ApoB Levels Be?
The target levels for apolipoprotein B (apoB) are a topic of ongoing debate in the medical community. ApoB is a critical marker because it represents the total number of atherogenic lipoprotein particles, which include LDL, VLDL, IDL, and Lp(a). These particles are closely associated with the risk of atherosclerosis and cardiovascular disease.
Aggressive Target Levels
Some experts advocate for very aggressive apoB targets, suggesting that the optimal levels should be similar to those found in young children, which are typically between 30-40 mg/dL. This aggressive approach aims to minimize the risk of atherosclerotic cardiovascular disease (ASCVD) as much as possible.
Guidelines and Recommendations
Different guidelines provide varying targets for apoB levels based on the patient's risk profile:
- Canadian Cardiovascular Society (CCS): The CCS recommends considering apoB levels in conjunction with LDL-C and non-HDL-C for risk assessment. They suggest a target apoB level of less than 0.7 g/L (70 mg/dL) for patients with established cardiovascular disease.
- American College of Cardiology (ACC) and American Heart Association (AHA): These organizations acknowledge apoB as a useful marker and often suggest it can be more reliable than LDL-C alone, particularly for high-risk patients.
So we've now reviewed how apoB burden can increase the risk of cardiovascular disease. Let's review the other major lipoprotein that poses a potential risk—Lp(a).
Understanding Lipoprotein(a) and Its Impact on Cardiovascular Health
Now let’s turn our attention to the other lipoprotein that is foundational to understanding your risk of cardiovascular disease—lipoprotein(a).
Lipoprotein(a), commonly referred to as Lp(a), is a lipoprotein particle that is garnering increasing attention within the medical community for its significant role in cardiovascular health. Despite its potentially lethal impact, it remains largely unknown to the general public. We’re going to outline why it is considered one of the most dangerous particles in the bloodstream and what can be done to manage its risks.
Lp(a) is structurally similar to low-density lipoprotein (LDL), which is commonly referred to as "bad cholesterol." However, Lp(a) includes a unique component that significantly alters its impact on cardiovascular health.
Lp(a) contains an additional protein called apolipoprotein(a), which is covalently attached to the apolipoprotein B-100 on the LDL particle. This addition not only distinguishes Lp(a) from LDL but also imparts different physiological and pathophysiological properties to the lipoprotein [33].
If Lp(a) is so potentially dangerous, why do we have it?
Evolutionary Benefit of Lp(a)
The evolutionary benefit of Lp(a) likely stems from its role in enhancing survival in ancient environments where trauma and bleeding were significant concerns. Here’s a detailed look at the potential evolutionary advantages:
Hypercoagulability
One of the key evolutionary advantages of Lp(a) is its role in blood clotting. Lp(a) closely resembles plasminogen, an essential protein in the fibrinolytic system, which is involved in breaking down blood clots. However, Lp(a) competes with plasminogen for binding sites, effectively inhibiting fibrinolysis (the process that breaks down clots) and promoting clot stability.
In pre-modern settings where minor injuries could lead to life-threatening blood loss, the hypercoagulable state conferred by higher Lp(a) levels would have been highly advantageous. During childbirth, hunting, or tribal warfare, enhanced clotting capability would help reduce hemorrhage risks, thereby increasing survival rates [34].
Scavenging Oxidized Lipids
Lp(a) also plays a role in the body’s handling of inflammation and oxidative stress. Lp(a) is effective at binding to oxidized phospholipids, which are known to play a role in the development of atherosclerosis and other inflammatory processes. By binding these oxidized lipids, Lp(a) helps remove them from circulation and arterial walls, potentially reducing oxidative damage and inflammation.
In environments where dietary sources and lifestyle did not predispose humans to chronic inflammation and oxidative stress (as seen in modern lifestyles with high processed food intake and sedentary habits), Lp(a)'s ability to scavenge oxidized lipids would have provided a protective benefit, helping to maintain cardiovascular and overall health [35].
Implications for Modern Health
While these traits may have been beneficial in ancient environments, in modern contexts, the evolutionary benefits of Lp(a) can translate into health risks.
In today's world, where prolonged life expectancy and different lifestyle factors come into play, the same properties of Lp(a) that once provided survival advantages can contribute to cardiovascular diseases. The hypercoagulability associated with high Lp(a) levels can lead to an increased risk of thrombosis and atherosclerosis, conditions that are major health concerns today.
Let's dive more into the potential risks of elevated Lp(a) and the underlying mechanisms of how those risks manifest.
The Dangers of Lipoprotein(a) (Lp(a))
Lp(a), possesses several hazardous properties that contribute significantly to cardiovascular risk. Here’s a detailed look at the primary hazardous properties of Lp(a):
Pro-atherogenic Properties of Lp(a)
At its core, Lp(a) contains an LDL particle, which is already known for its role in atherosclerosis. LDL particles carry cholesterol into the arterial wall, where they can become trapped and oxidized, forming the basis of atherosclerotic plaques. The presence of LDL within Lp(a) means that it shares these inherent atherogenic properties.
Similar to LDL, Lp(a) can undergo oxidation once it penetrates the arterial wall. Oxidized Lp(a) is particularly problematic because it triggers inflammation, attracts immune cells, and can contribute to the formation of the fatty streaks that are precursors to more mature atherosclerotic plaques. This oxidation process heightens the particle’s atherogenicity and is a critical step in plaque development [35].
Pro-inflammatory Properties
Lp(a) is known to transport oxidized phospholipids, which are highly inflammatory molecules. These oxidized phospholipids play a crucial role in the pathogenesis of atherosclerosis because they can activate endothelial cells and recruit immune cells such as monocytes and macrophages to the site. The accumulation of these cells and their transformation into foam cells contribute significantly to the development and progression of atherosclerotic plaques. This recruitment is a critical step in the transformation of early atherosclerotic lesions into more complex, unstable plaques that are prone to rupture, leading to acute cardiovascular events [35].
The presence of Lp(a) in the arterial wall is associated with a chronic inflammatory response. This is particularly detrimental because chronic inflammation is a key driver in the progression of atherosclerosis. Inflammatory processes foster the continuous growth of plaques and contribute to their vulnerability to rupture. Lp(a) not only initiates but also perpetuates inflammation within the arterial wall, thereby enhancing the risk of plaque destabilization and potential thrombotic complications [3].
Pro-thrombotic Properties of Lp(a)
Lp(a) plays a significant role in increasing the risk of thrombosis due to its unique molecular structure and interactions.
Lp(a) is structurally similar to plasminogen, a key enzyme in the fibrinolytic system responsible for breaking down fibrin clots. The critical aspect of Lp(a)’s structure is its apolipoprotein(a) component, which closely resembles plasminogen. This resemblance allows Lp(a) to bind to fibrin and other receptors that normally bind plasminogen, effectively competing for these binding sites.
By competing with plasminogen for binding sites on fibrin, Lp(a) inhibits the normal fibrinolytic process. Fibrinolysis is the biological breakdown of blood clots, and its inhibition by Lp(a) means that clots are less likely to be dissolved once formed. This inhibition not only promotes the stability and persistence of existing clots but also facilitates the formation of new thrombi [34].
The pro-thrombotic properties of Lp(a) have direct and severe implications for cardiovascular health, particularly in the formation of acute thrombotic events.
The ability of Lp(a) to interfere with clot breakdown directly contributes to an increased risk of conditions such as heart attacks and strokes. In scenarios where a clot forms within a coronary or cerebral artery, the impaired fibrinolytic activity due to Lp(a) can lead to sustained blockages, severely compromising blood flow and leading to tissue damage or organ failure.
The persistence of clots due to Lp(a)'s antagonistic action on fibrinolysis can lead to chronic cardiovascular conditions and complications. For example, persistent clots in the coronary arteries can lead to chronic coronary artery disease, while in peripheral arteries, they can cause peripheral artery disease.
Contribution of Lp(a) to Aortic Stenosis
Lp(a) plays a significant role in the pathogenesis of aortic stenosis, a serious cardiovascular condition characterized by the narrowing of the aortic valve, through its effects on calcification and cellular activities within the aortic valve.
One of the primary mechanisms by which Lp(a) contributes to aortic stenosis is through the calcification of the aortic valve.
High levels of Lp(a) are associated with increased deposition of calcium on the valve. This calcification process is not merely a passive accumulation but is actively influenced by the lipoproteins carried by Lp(a), which can modify the extracellular matrix and cellular responses within the valve tissue. The calcified valve becomes less compliant and more narrowed, restricting blood flow from the heart into the aorta, which can lead to heart failure if left untreated [36].
Lp(a) also affects the cellular dynamics within the aortic valve by stimulating the production of bone-forming proteins, such as osteopontin and bone morphogenetic proteins. These proteins contribute to the osteogenic transformation of valvular interstitial cells, leading to the stiffening and narrowing of the aortic valve. This process exacerbates the symptoms of aortic stenosis, as the valve becomes increasingly unable to open fully, thereby obstructing blood flow and increasing the heart's workload [36].
Here is the important point: Individuals with high Lp(a) levels face 2 to 4 times the risk of developing aortic stenosis [36].
Lp(a) and Increased Risk of Venous Thromboembolism
Lp(a) is primarily known for its role in atherosclerosis and cardiovascular diseases, but it also plays a significant part in venous thrombotic events.
Elevated Lp(a) levels have been correlated with an increased risk of venous thromboembolism. Venous thromboembolism encompasses both deep vein thrombosis, which typically occurs in the deep veins of the legs, and pulmonary embolism, where clot fragments travel to the pulmonary arteries. These conditions are significant health risks as they can lead to severe complications, including sudden death in the case of significant pulmonary embolism.
The exact mechanisms by which Lp(a) contributes to VTE are not fully understood but are believed to involve its pro-thrombotic properties. Lp(a)’s structural similarity to plasminogen, as previously noted, allows it to interfere with the body's natural fibrinolytic system, thereby promoting clot stability and persistence. This interference can predispose individuals to thrombus formation not only in the arterial system but also in the venous system [37].
What to know about the risk of VTE when you have elevated Lp(a): The hazard ratio for VTE is around 3, indicating a tripled risk for those with elevated Lp(a) levels [37]
Lp(a) Hazard Ratios and Atherosclerosis Risk
Via its pro-atherogenic properties, pro-inflammatory actions, and pro-thrombotic effects LP(a)’s impact on the risk of developing atherosclerosis is significant.
The hazard ratios associated with Lp(a) levels for atherosclerosis range from 2 to 4, which means that individuals with elevated Lp(a) levels have two to four times the risk of developing atherosclerosis compared to those with normal Lp(a) levels. This substantial increase highlights the potent atherogenic nature of Lp(a).
Specifically, some studies have shown that high Lp(a) concentrations can result in a 60% increased risk of atherosclerosis. This indicates a significant escalation in risk, underscoring the importance of Lp(a) as a critical factor in the development of this condition [35].
Genetic Predisposition to Elevated Lp(a) Levels
One of the most challenging aspects of lipoprotein(a) [Lp(a)] is that its levels are largely determined by genetics. Unlike low-density lipoprotein (LDL) cholesterol, which can be significantly influenced by diet and lifestyle, Lp(a) levels are primarily inherited. This means that individuals with high Lp(a) levels often have a family history of cardiovascular disease. It is estimated that about 20% of the global population has elevated Lp(a) levels, making it a significant, yet often overlooked, risk factor [38].
Despite its dangers, Lp(a) is not commonly measured in routine lipid panels. This oversight can leave many individuals unaware of their heightened cardiovascular risk. Increasing awareness about Lp(a) and advocating for its inclusion in standard lipid profiles is essential. A simple blood test can determine Lp(a) levels, providing critical information for risk assessment and management [39].
Genetic testing can identify individuals at risk due to high Lp(a) levels, allowing for early intervention. Family history plays a crucial role in predicting elevated Lp(a) levels, and genetic screening can help identify those who might benefit from more aggressive management of other cardiovascular risk factors [40].
Current and Emerging Treatments for Elevated Lp(a) Levels
Despite the significant cardiovascular risks associated with high Lp(a) levels, traditional lipid-lowering therapies like statins typically do not affect Lp(a). This has led to the exploration of alternative treatments. Statins, while effective at lowering LDL cholesterol, have little to no impact on Lp(a) levels. This limitation necessitates alternative strategies specifically targeting Lp(a).
Role of Niacin in Managing Lp(a) – (It probably can’t hurt)
Niacin, also known as vitamin B3, has been utilized to manage lipid profiles, including reducing Lp(a) levels. Niacin can decrease Lp(a) levels by about 20-30%. It achieves this reduction primarily by inhibiting the secretion of apolipoprotein(a) from the liver, which is crucial for the formation of Lp(a) particles [41].
Niacin has been used to manage lipid levels, including Lp(a). Here's how niacin helps with elevated Lp(a) and its overall effect on lipid profiles:
Reduction of Lp(a) Levels: Niacin can reduce Lp(a) levels by approximately 20-30%. Niacin's primary action in reducing Lp(a) levels is its ability to inhibit the secretion of apolipoprotein(a) from the liver. As we have mentioned, apolipoprotein(a) is an integral component of Lp(a) particles. By decreasing its secretion, niacin effectively reduces the synthesis of new Lp(a) particles, leading to lower circulating levels of Lp(a) [41].
Reduction in LDL-C: Niacin can decrease LDL-C levels by 5% to 25%. This range indicates variability in response, which may depend on the dosage and the baseline lipid levels of the patient [41].
Reduction in Triglycerides: Niacin is particularly effective in lowering triglyceride levels, with reductions ranging from 20% to 50%. This significant decrease in triglycerides also contributes to the overall reduction in VLDL levels, impacting LDL-C [41]..
Alteration of LDL Particle Composition: Niacin also affects the composition of LDL particles, shifting their distribution from small, dense LDL particles, which are more atherogenic, to larger, more buoyant particles. These larger particles are considered less harmful and are associated with a reduced risk of CHD [41].
Mechanism of Action of Niacin on Lipid Metabolism
Niacin has several mechanisms that contribute to its lipid-modifying effects, particularly its ability to reduce LDL-C and triglyceride levels:
Reduction of VLDL Synthesis: Niacin decreases the mobilization of free fatty acids from adipose tissue. Lower levels of free fatty acids lead to reduced hepatic synthesis of triglycerides, which are key components of VLDL. Since VLDL is a precursor to LDL, reducing VLDL synthesis indirectly leads to lower LDL-C levels [41].
Inhibition of Apolipoprotein B-100: Niacin inhibits the synthesis of apolipoprotein B-100, a protein essential for the assembly of VLDL particles. By inhibiting this protein, niacin effectively reduces the production of VLDL particles, thereby impacting LDL-C levels [41].
Enhancement of VLDL Catabolism: Niacin induces the activity of lipoprotein lipase, an enzyme that plays a critical role in the catabolism of VLDL. Increased lipoprotein lipase activity enhances the breakdown of VLDL, which also contributes to lower levels of LDL-C [41].
Anti-Inflammatory Properties: Beyond its lipid-altering actions, niacin also exhibits significant anti-inflammatory effects. It can reduce the inflammation associated with atherosclerotic plaques. Chronic inflammation plays a pivotal role in the progression of atherosclerosis, contributing to plaque instability and the potential for acute cardiovascular events, such as myocardial infarction and stroke. By mitigating inflammatory processes within the arterial wall, niacin contributes further to cardiovascular protection [42].
Tempering Expectations Around Niacin Use
Despite the established benefits of niacin in improving lipid profiles, its role in cardiovascular risk management has been subject to debate due to mixed results from significant clinical trials.
AIM-HIGH and HPS2-THRIVE Trials
These major clinical trials critically influenced the perception of niacin's effectiveness in cardiovascular disease prevention. Both studies aimed to assess the impact of adding niacin to statin therapy on cardiovascular outcomes. However, they failed to demonstrate a significant reduction in major cardiovascular events, which has led to questions about the value of niacin in routine clinical practice when used in combination with statins.The AIM-HIGH trial specifically questioned the additional benefit of niacin in patients already well-controlled on statins, finding no significant reduction in cardiovascular events [43].
The HPS2-THRIVE study similarly found that niacin did not significantly reduce the risk of major vascular events and highlighted significant side effects associated with niacin therapy [44].
However, there were major flaws in these studies. Overall, niacin use has very little downside and enough data to consider utilizing it as a way to reduce overall risk of cardiovascular disease.
Statins and Their Impact on Lp(a) Levels (None)
Statins, widely used to lower LDL cholesterol and reduce cardiovascular risk, have a more complex and less effective relationship with Lp(a) levels. Statins generally do not significantly reduce Lp(a) levels.
Unlike their significant impact on LDL cholesterol, statins generally do not substantially reduce Lp(a) levels. The primary mechanisms by which statins reduce cholesterol levels—mainly through the reduction of cholesterol synthesis and the upregulation of LDL receptors—do not effectively target the metabolic pathways involved in Lp(a) production or clearance.
Interestingly, statin therapy has been observed to slightly increase Lp(a) levels in some individuals. This effect may be linked to the upregulation of hepatic LDL receptors, which, while increasing the clearance of LDL cholesterol, does not similarly enhance the clearance of Lp(a). This phenomenon could potentially be due to the unique structural components of Lp(a) that affect its interaction with LDL receptors [45].
Understanding how statins work helps clarify why their impact on Lp(a) is limited. Statins primarily inhibit the enzyme HMG-CoA reductase in the liver. This enzyme is crucial for cholesterol synthesis. By inhibiting this pathway, statins reduce the overall synthesis of cholesterol. This reduction leads to an upregulation of LDL receptors on liver cells as the body attempts to compensate for lower cholesterol levels. The increased number of LDL receptors enhances the clearance of LDL particles from the bloodstream, effectively lowering LDL cholesterol levels [46].
While the increase in LDL receptors also affects Lp(a), the clearance of Lp(a) by these receptors is not as efficient as that of LDL. This inefficiency is due to the structural differences between LDL and Lp(a), particularly the presence of the apolipoprotein(a) component in Lp(a), which influences how Lp(a) interacts with LDL receptors. Therefore, despite the increase in LDL receptors induced by statins, the clearance of Lp(a) does not improve significantly [35].
PCSK9 Inhibitors: Mechanism and Clinical Benefits (Reduces Lp(a))
Mechanism of Action of PCSK9 Inhibitors
PCSK9 inhibitors are a relatively new class of drugs that have transformed the management of patients with high cholesterol, especially those who are at high risk of cardiovascular events or those who do not respond adequately to statins.
PCSK9 binds to LDL receptors on the surface of liver cells, promoting their degradation. LDL receptors are crucial for removing LDL cholesterol from the bloodstream. When these receptors are degraded, the liver’s capacity to clear LDL cholesterol is diminished, leading to higher blood levels of LDL cholesterol [32].
PCSK9 inhibitors, which are monoclonal antibodies, target and bind to PCSK9. By inhibiting PCSK9, these drugs prevent it from binding to LDL receptors. This action prevents the receptors from being degraded, thereby increasing their availability on the liver cell surface. More LDL receptors lead to enhanced clearance of LDL cholesterol from the blood.
This is important: PCSK9 Inhibitors lead to the clearance of LDL. This has implications for LP(a) as well.
Effects on LDL
The increased availability of LDL receptors due to PCSK9 inhibition results in significantly lower levels of LDL cholesterol in the bloodstream. Clinical studies have shown that PCSK9 inhibitors can reduce LDL cholesterol levels by 50-70%, which is a substantial decrease, particularly beneficial for patients who are at high risk of cardiovascular diseases or those who cannot achieve their LDL targets with statins alone [32].
Effects on Lp(a) Levels
In addition to lowering LDL cholesterol, PCSK9 inhibitors also reduce levels of Lp(a) by about 20-30%. This reduction is particularly noteworthy because Lp(a) is a significant risk factor for cardiovascular diseases, and there are few effective treatments available that specifically lower Lp(a) levels. The mechanism here is similar to that for LDL — by increasing LDL receptor availability, PCSK9 inhibitors also enhance the clearance of Lp(a) particles from the bloodstream [35].
The degree of Lp(a) reduction can vary among individuals. Some patients may experience more significant reductions, while others may have a more modest response.
The combined reduction of LDL cholesterol and Lp(a) levels by PCSK9 inhibitors contributes to a lower overall cardiovascular risk. Clinical trials have demonstrated that these medications significantly reduce the incidence of major cardiovascular events such as heart attacks and strokes in high-risk patients [32].
Antisense Oligonucleotides (ASOs): A Promising Approach to Lowering Lp(a) Levels in Clinical Trials
ASOs are a novel class of therapeutics that modulate gene expression at the mRNA level. ASOs are specially designed, short synthetic strands of nucleotides that specifically bind to the messenger RNA (mRNA) encoding apolipoprotein(a) [Apo(a)], which is a key protein component of Lp(a). By binding to Apo(a) mRNA, ASOs block its translation process, thereby preventing the synthesis of the Apo(a) protein itself [32].
The inhibition of Apo(a) synthesis by ASOs leads to a decrease in the assembly and secretion of Lp(a) particles from the liver. Consequently, this action results in lowered circulating levels of Lp(a) in the bloodstream, effectively reducing one of the major risk factors for cardiovascular diseases [32].
The clinical trials involving ASOs have shown promising results in terms of their capacity to lower Lp(a) levels. Clinical studies have documented that ASOs can reduce Lp(a) levels by an impressive 80-90%. This dramatic decrease can potentially normalize Lp(a) levels in patients who previously had elevated concentrations, thus mitigating associated risks [47].
Important Point: While ASOs have much potential, they are currently in a clinical trial phase and are not commonly used for Lp(a) reduction.
Conclusion
In conclusion, this research review has illuminated a pivotal shift in the understanding of cardiovascular health, moving from a traditional focus on cholesterol to a more precise consideration of lipoprotein behavior, especially apoB and Lp(a). The catalyst for this shift was Dr. Allan Sniderman's critical narrative review in JAMA Cardiology, which introduced the ApoB Particle Model of Atherogenesis. This model challenges conventional cholesterol metrics, advancing apoB particle count as a more accurate predictor of cardiovascular risk, thereby transforming our approach to cardiovascular prevention and treatment.
Through a detailed exploration of the mechanisms by which cholesterol becomes harmful, particularly under the influence of metabolic dysfunctions like elevated insulin levels and mitochondrial dysfunction, we have seen how atherosclerosis is not merely about the presence of cholesterol but more critically about the behavior of specific lipoproteins in the vascular environment.
The clinical implications of the ApoB Particle Model are profound, suggesting that reducing apoB particles can significantly decrease the risk of heart disease. This review also addressed the substantial risks posed by Lp(a), detailing its evolutionary background, modern health implications, and the strategies needed to mitigate its impact.
By concluding with actionable strategies for managing levels of Lp(a) and apoB, this review does not merely expand our understanding of cardiovascular risk factors but actively provides guidance on reducing these risks through lifestyle modifications and targeted therapeutic interventions. As we bridge the gap between research findings and clinical practice, the goal is to empower healthcare professionals and patients alike to more effectively combat cardiovascular disease, ensuring a proactive approach to heart health that is informed by the latest scientific insights.
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007 Oct 16;116(16):1832-44. doi: 10.1161/CIRCULATIONAHA.106.676890. PMID: 17938300.
- Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009 Apr;50 Suppl(Suppl):S376-81. doi: 10.1194/jlr.R800087-JLR200. Epub 2008 Nov 15. PMID: 19011257; PMCID: PMC2674707.
- Libby, P., Ridker, P. & Hansson, G. Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 (2011). https://doi.org/10.1038/nature10146
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999 Jan 14;340(2):115-26. doi: 10.1056/NEJM199901143400207. PMID: 9887164.
- Saltiel, A., Kahn, C. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 (2001). https://doi.org/10.1038/414799a
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988 Dec;37(12):1595-607. doi: 10.2337/diab.37.12.1595. PMID: 3056758.
- Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002 Apr;23(2):201-29. doi: 10.1210/edrv.23.2.0461. PMID: 11943743.
- Adiels M, Olofsson SO, Taskinen MR, Borén J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008 Jul;28(7):1225-36. doi: 10.1161/ATVBAHA.107.160192. PMID: 18565848.
- Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996 Mar;19(3):257-67. doi: 10.2337/diacare.19.3.257. PMID: 8742574.
- Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006 Apr 4;113(13):1708-14. doi: 10.1161/CIRCULATIONAHA.105.602532. PMID: 16585403.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006 Dec 14;444(7121):860-7. doi: 10.1038/nature05485. PMID: 17167474.
- Libby, P. (2002). Inflammation in atherosclerosis. Nature, 420(6917), 868-874.
- Joseph L. Evans, Ira D. Goldfine, Betty A. Maddux, Gerold M. Grodsky, Oxidative Stress and Stress-Activated Signaling Pathways: A Unifying Hypothesis of Type 2 Diabetes, Endocrine Reviews, Volume 23, Issue 5, 1 October 2002, Pages 599–622, https://doi.org/10.1210/er.2001-0039
- Rask-Madsen C, King GL. Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007 Jan;3(1):46-56. doi: 10.1038/ncpendmet0366. PMID: 17179929.
- Colberg SR, Sigal RJ, Fernhall B, Regensteiner JG, Blissmer BJ, Rubin RR, Chasan-Taber L, Albright AL, Braun B; American College of Sports Medicine; American Diabetes Association. Exercise and type 2 diabetes: the American College of Sports Medicine and the American Diabetes Association: joint position statement. Diabetes Care. 2010 Dec;33(12):e147-67. doi: 10.2337/dc10-9990. PMID: 21115758; PMCID: PMC2992225.
- Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007 Mar 2;100(4):460-73. doi: 10.1161/01.RES.0000258450.44413.96. PMID: 17332437.
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012 Mar;122(6):253-70. doi: 10.1042/CS20110386. PMID: 22117616; PMCID: PMC3398862.
- Mahley RW, Innerarity TL. Lipoprotein receptors and cholesterol homeostasis. Biochim Biophys Acta. 1983 May 24;737(2):197-222. doi: 10.1016/0304-4157(83)90001-1. PMID: 6303423.
- Hevonoja T, Pentikäinen MO, Hyvönen MT, Kovanen PT, Ala-Korpela M. Structure of low density lipoprotein (LDL) particles: basis for understanding molecular changes in modified LDL. Biochim Biophys Acta. 2000 Nov 15;1488(3):189-210. doi: 10.1016/s1388-1981(00)00123-2. PMID: 11082530.
- Feingold, K.R., & Grunfeld, C. (2000). Role of cytokines in inducing hyperlipidemia. Diabetes, 39(Suppl 2), 3-5.
- Ginsberg HN. Lipoprotein physiology. Endocrinol Metab Clin North Am. 1998 Sep;27(3):503-19. doi: 10.1016/s0889-8529(05)70023-2. PMID: 9785050.
- HAVEL RJ, EDER HA, BRAGDON JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955 Sep;34(9):1345-53. doi: 10.1172/JCI103182. PMID: 13252080; PMCID: PMC438705.
- Sniderman AD, Thanassoulis G, Glavinovic T, Navar AM, Pencina M, Catapano A, Ference BA. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019 Dec 1;4(12):1287-1295. doi: 10.1001/jamacardio.2019.3780. PMID: 31642874; PMCID: PMC7369156.
- Sniderman AD, St-Pierre AC, Cantin B, Dagenais GR, Després JP, Lamarche B. Concordance/discordance between plasma apolipoprotein B levels and the cholesterol indexes of atherosclerotic risk. Am J Cardiol. 2003 May 15;91(10):1173-7. doi: 10.1016/s0002-9149(03)00262-5. PMID: 12745098.
- Ahmad M, Sniderman AD, Hegele RA. Apolipoprotein B in cardiovascular risk assessment. CMAJ. 2023 Aug 28;195(33):E1124. doi: 10.1503/cmaj.230048. PMID: 37640407; PMCID: PMC10462411.
- Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008 Jan;54(1):24-38. doi: 10.1373/clinchem.2007.097360. PMID: 18160725.
- Krauss RM. Lipoprotein subfractions and cardiovascular disease risk. Curr Opin Lipidol. 2010 Aug;21(4):305-11. doi: 10.1097/MOL.0b013e32833b7756. PMID: 20531184.
- Williams, K.J., & Tabas, I. (1995). The response-to-retention hypothesis of early atherogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology, 15(5), 551-561.
- Schiopu A, Bengtsson J, Söderberg I, Janciauskiene S, Lindgren S, Ares MP, Shah PK, Carlsson R, Nilsson J, Fredrikson GN. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation. 2004 Oct 5;110(14):2047-52. doi: 10.1161/01.CIR.0000143162.56057.B5. Epub 2004 Sep 27. PMID: 15451805.
- Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, Laufs U, Oliver-Williams C, Wood AM, Butterworth AS, Di Angelantonio E, Danesh J, Nicholls SJ, Bhatt DL, Sabatine MS, Catapano AL. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019 Jan 29;321(4):364-373. doi: 10.1001/jama.2018.20045. PMID: 30694319; PMCID: PMC6439767.
- Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM; IMPROVE-IT Investigators. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015 Jun 18;372(25):2387-97. doi: 10.1056/NEJMoa1410489. Epub 2015 Jun 3. PMID: 26039521.
- Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR; FOURIER Steering Committee and Investigators. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017 May 4;376(18):1713-1722. doi: 10.1056/NEJMoa1615664. Epub 2017 Mar 17. PMID: 28304224.
- Vinci P, Di Girolamo FG, Panizon E, Tosoni LM, Cerrato C, Pellicori F, Altamura N, Pirulli A, Zaccari M, Biasinutto C, Roni C, Fiotti N, Schincariol P, Mangogna A, Biolo G. Lipoprotein(a) as a Risk Factor for Cardiovascular Diseases: Pathophysiology and Treatment Perspectives. Int J Environ Res Public Health. 2023 Sep 6;20(18):6721. doi: 10.3390/ijerph20186721. PMID: 37754581; PMCID: PMC10531345.
- Berglund L, Ramakrishnan R. Lipoprotein(a): an elusive cardiovascular risk factor. Arterioscler Thromb Vasc Biol. 2004 Dec;24(12):2219-26. doi: 10.1161/01.ATV.0000144010.55563.63. Epub 2004 Sep 2. PMID: 15345512; PMCID: PMC3275911.
- Tsimikas S. Oxidative biomarkers in the diagnosis and prognosis of cardiovascular disease. Am J Cardiol. 2006 Dec 4;98(11A):9P-17P. doi: 10.1016/j.amjcard.2006.09.015. Epub 2006 Sep 26. PMID: 17126679.
- Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014 Feb 11;63(5):470-7. doi: 10.1016/j.jacc.2013.09.038. Epub 2013 Oct 23. PMID: 24161338.
- Dentali F, Gessi V, Marcucci R, Gianni M, Grandi AM, Franchini M. Lipoprotein(a) as a Risk Factor for Venous Thromboembolism: A Systematic Review and Meta-analysis of the Literature. Semin Thromb Hemost. 2017 Sep;43(6):614-620. doi: 10.1055/s-0036-1598002. Epub 2017 Mar 27. PMID: 28346964.
- Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992 Jul;90(1):52-60. doi: 10.1172/JCI115855. PMID: 1386087; PMCID: PMC443062.
- Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010 Dec;31(23):2844-53. doi: 10.1093/eurheartj/ehq386. Epub 2010 Oct 21. PMID: 20965889; PMCID: PMC3295201.
- Clarke, R., Peden, J.F., Hopewell, J.C., Kyriakou, T., Goel, A., Heath, S.C., ... & Farrall, M. (2009). Genetic variants associated with Lp(a) lipoprotein level and coronary disease. New England Journal of Medicine, 361(26), 2518-2528.
- McKenney J. New Perspectives on the Use of Niacin in the Treatment of Lipid Disorders. Arch Intern Med. 2004;164(7):697–705. doi:10.1001/archinte.164.7.697
- Ganji SH, Kamanna VS, Kashyap ML. Niacin and cholesterol: role in cardiovascular disease (review). J Nutr Biochem. 2003 Jun;14(6):298-305. doi: 10.1016/s0955-2863(02)00284-x. PMID: 12873710.
- AIM-HIGH Investigators; Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011 Dec 15;365(24):2255-67. doi: 10.1056/NEJMoa1107579. Epub 2011 Nov 15. Erratum in: N Engl J Med. 2012 Jul 12;367(2):189. PMID: 22085343.
- HPS2-THRIVE Collaborative Group; Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014 Jul 17;371(3):203-12. doi: 10.1056/NEJMoa1300955. PMID: 25014686.
- Burgess S, Ference BA, Staley JR, Freitag DF, Mason AM, Nielsen SF, Willeit P, Young R, Surendran P, Karthikeyan S, Bolton TR, Peters JE, Kamstrup PR, Tybjærg-Hansen A, Benn M, Langsted A, Schnohr P, Vedel-Krogh S, Kobylecki CJ, Ford I, Packard C, Trompet S, Jukema JW, Sattar N, Di Angelantonio E, Saleheen D, Howson JMM, Nordestgaard BG, Butterworth AS, Danesh J; European Prospective Investigation Into Cancer and Nutrition–Cardiovascular Disease (EPIC-CVD) Consortium. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018 Jul 1;3(7):619-627. doi: 10.1001/jamacardio.2018.1470. PMID: 29926099; PMCID: PMC6481553.
- Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res. 1992 Nov;33(11):1569-82. PMID: 1464741.
- Viney, N.J., van Capelleveen, J.C., Geary, R.S., Xia, S., Tami, J.A., Yu, R., ... & Crooke, R.M. (2016). Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. The Lancet, 388(10057), 2239-2253.
Related studies